



Содержание
Перейти к:
 А. С. Петросян,
Е. Е. Бибик,
Р. Х. Салимханов,
Е. В. Ковалева,
А. К. Еремкина,
М. В. Уткина,
Е. А. Трошина,
Н. Г. Мокрышева
А. С. Петросян,
Е. Е. Бибик,
Р. Х. Салимханов,
Е. В. Ковалева,
А. К. Еремкина,
М. В. Уткина,
Е. А. Трошина,
Н. Г. Мокрышева https://doi.org/10.14341/osteo13183
Перейти к:
Синдром множественных эндокринных неоплазий 4 типа (МЭН-4) — редкое аутосомно-доминантное заболевание, возникающее в результате мутации в гене CDKN1B, кодирующем регулятор клеточного цикла p27. В связи с редкостью патологии к настоящему времени собрано небольшое количество клинических наблюдений за пациентами с мутациями CDKN1B, при этом наличие генотип-фенотипических корреляций остается дискутабельным и требует дополнительного уточнения. При МЭН-4 в патологический процесс вовлекаются те же органы, что и при МЭН-1, однако возраст манифестации и течение заболевания могут отличаться. Мы представляем описание клинического случая пациентки с МЭН-4 с ранее не описанной в литературе мутацией в гене CDKN1B со сдвигом рамки считывания, которая привела к развитию первичного гиперпаратиреоза (ПГПТ) с множественным поражением околощитовидных желез (ОЩЖ) и пролактин-секретирующей микроаденомы гипофиза. Первым компонентом заболевания, диагностированным в возрасте 37 лет, была пролактинома. Позднее была выявлена висцеральная форма ПГПТ с изолированным поражением почек. Пациентке проводилось специфическое лечение указанных патологий с достижением удовлетворительных результатов. При комплексном обследовании значимого поражения других органов выявлено не было. Ограниченное число наблюдений за пациентами с мутациями в гене CDKN1B в настоящее время не позволяет определить закономерности течения этого заболевания, в связи с чем подробное описание клинической картины при новой выявленной мутации вносит значимый вклад в изучение данной патологии.
Петросян А.С., Бибик Е.Е., Салимханов Р.Х., Ковалева Е.В., Еремкина А.К., Уткина М.В., Трошина Е.А., Мокрышева Н.Г. Новая гетерозиготная мутация в гене CDKN1B у пациентки с синдромом множественных эндокринных неоплазий 4 типа. Остеопороз и остеопатии. 2024;27(4):31-37. https://doi.org/10.14341/osteo13183
Petrosyan A.S., Bibik E.E., Salimkhanov R.H., Kovaleva E.V., Eremkina A.K., Utkina M.V., Troshina E.A., Mokrysheva N.G. A new heterozygous mutation in the CDKN1B gene in a patient affected by multiple endocrine neoplasia syndrome type 4. Osteoporosis and Bone Diseases. 2024;27(4):31-37. (In Russ.) https://doi.org/10.14341/osteo13183
Синдром множественных эндокринных неоплазий 4 типа (МЭН-4) — редкое аутосомно-доминантное заболевание, возникающее в результате герминативной мутации в гене CDKN1B, кодирующем регулятор клеточного цикла p27. Синдром МЭН-4 ассоциирован с первичным гиперпаратиреозом (ПГПТ), аденомами гипофиза, нейроэндокринными нейроэндокринными опухолями (НЭО) желудочно-кишечного тракта (ЖКТ), поджелудочной железы и другими новообразованиями, являясь фенотипической копией синдрома множественных эндокринных неоплазий 1 типа (МЭН-1). В связи с редкостью патологии к настоящему времени собрано небольшое количество клинических наблюдений за пациентами с мутациями CDKN1B, при этом наличие генотип-фенотипических корреляций при МЭН-4 остается дискутабельным и требует дополнительного уточнения. На 2024 г. в литературе описано лишь 65 пациентов с мутацией в гене, кодирующем p27. Среди них было обнаружено 28 различных вариантов CDKN1B, большинство из которых являлись миссенс-мутациями или мутациями сдвига рамки считывания [1].
Известно, что при МЭН-4 в патологический процесс вовлекаются те же органы, что и при МЭН-1, однако возраст манифестации и течение заболевания могут отличаться. Мутация в гене CDKN1B приводит к развитию ПГПТ в 75–90% случаев (чаще гистологически представленного солитарными аденомами околощитовидных желез (ОЩЖ)), в 40–44% диагностируются аденомы гипофиза, а в 19–25% — панкреато-дуоденальные НЭО. Реже у таких пациентов встречаются папиллярный рак щитовидной железы (ПРЩЖ), новообразования надпочечников, тимуса [1][2]. В описанных случаях первые проявления синдрома МЭН-4 возникали в среднем возрасте, чаще у женщин, чем у мужчин, а пик заболеваемости приходился на пятую декаду жизни [1].
Мы представляем описание клинического случая пациентки с МЭН-4 с ранее не описанной в литературе мутацией в гене CDKN1B со сдвигом рамки считывания, которая привела к развитию ПГПТ с множественным поражением ОЩЖ и пролактин-секретирующей микроаденомы гипофиза.
Пациентка С., 57 лет, госпитализирована в отделение патологии ОЩЖ и нарушений минерального обмена (ОПОЩЖ и НМО) ФГБУ «НМИЦ эндокринологии» Минздрава России в феврале 2023 г.
Из анамнеза известно, что около 20 лет назад, в возрасте 37 лет, впервые была диагностирована гиперпролактинемия до 3000 мМЕ/л (сведения об уровне биоактивного пролактина отсутствуют). Со слов пациентки, образований гипоталамо-гипофизарной области тогда выявлено не было. В течение двух лет проводилась терапия агонистом дофаминовых рецепторов (бромокриптином) с достижением нормопролактинемии. В дальнейшем в ходе нерегулярного динамического контроля отмечалось повышение уровня общего пролактина до 1000 мМЕ/л, лечение не назначалось. Менопауза у пациентки наступила в 48 лет после гистеросальпингооварэктомии по поводу миомы матки крупных размеров. При подробном расспросе особенностей наследственного анамнеза не отмечено.
Впервые повышение концентрации паратгормона (ПТГ) крови до 84,21 пг/мл при гиперкальциемии по кальцию общему 2,72–2,9 ммоль/л, гиперкальциурии 12,8 ммоль/сут, дефиците 25(ОН)витамина D — 10,6 нг/мл было выявлено за 6 месяцев до госпитализации, пациентке рекомендован прием колекальциферола в дозе 4000 МЕ в сутки.
При поступлении в стационар женщина предъявляла жалобы на боли в коленных и тазобедренных суставах, общую слабость, утомляемость, бессонницу. При осмотре телосложение правильное, рост 180 см, вес 92 кг (ИМТ 28,4 кг/м²), видимых деформаций скелета не отмечалось. Указаний на перенесенные переломы в анамнезе не было. В ходе обследования у пациентки был подтвержден ПГПТ (табл. 1). В рамках скрининга осложнений заболевания выявлены эхографические признаки пиелокаликоэктазии правой почки, двухстороннего нефромикролитиаза, по данным рентгеноденситометрии минеральная плотность костной ткани (МПК) по T-критерию в поясничном отделе позвоночника (L1-4) составила -1,6 SD, в лучевой и бедренной костях соответствовала нормальным значениям: 0,0 SD (Radius 33%), -0,9 SD (Femur Neck) и -0,3 SD (Total Hip). По результатам рентгенографии грудного и поясничного отделов позвоночника в боковой проекции компрессионные переломы тел позвонков не обнаружены. При УЗИ органов шеи визуализированы образования четырех ОЩЖ в типичных местах (правая верхняя размерами 1,2х0,9х0,6 см, правая нижняя 1,7х1,3х0,8 см, левая верхняя 1,4х0,8х0,5 см, левая нижняя 1,5х0,7х0,4 см). При этом, по результатам сцинтиграфии ОЩЖ (99mTc-Технетрил) с однофотонной эмиссионной компьютерной томографией (ОФЭКТ-КТ), накопление радиофармпрепарата наблюдалось только в проекции правой нижней ОЩЖ, что было расценено как снижение чувствительности исследования при полигландулярном поражении ОЩЖ.
Таблица 1. Показатели лабораторной диагностики пациентки С. за время наблюдения
|
Показатель Дата |
ПТГ, пг/мл (РИ 15–65) |
Кальций общий, ммоль/л (РИ 2,15–2,55) |
Кальций альбумин-скорректированный, ммоль/л (РИ 2,15–2,55) |
Фосфор, ммоль/л (РИ 0,74–1,52) |
Кальций суточной мочи, ммоль/сут (РИ 2,5–8,0) |
рСКФ по CKD-EPI, мл/мин/1,73 м² |
ТТГ, мЕд/л (РИ 0,25–3,50) |
ИФР-1, нг/мл (РИ 17–238) |
Пролактин общий, мЕд/л (РИ 69–340) |
|
Февраль 2023 г. (ЭНЦ) |
56,6 |
- |
2,65 |
- |
16,8 |
- |
- |
- |
796,6 |
|
Апрель 2023 г. (ЭНЦ), после ПТЭ |
3,9 |
2,24 |
- |
- |
- |
- |
- |
- |
- |
|
Декабрь 2023 г. |
- |
2,16 |
- |
- |
8,3 |
73 |
0,77 |
- |
- |
|
Март 2024 г. |
8,5 |
- |
2,26 |
1,06 |
- |
- |
1,85 |
- |
914 |
|
Июль 2024 г. (ЭНЦ) |
- |
- |
2,17 |
1,15 |
8,3 |
91 |
1,3 |
204,4 |
820,8 |
Примечание: ЭНЦ — Эндокринологический научный центр (ФГБУ «НМИЦ эндокринологии» Минздрава России), ПТГ — паратиреоидный гормон; ПТЭ — паратиреоидэктомия; РИ — референсный интервал; рСКФ по CKD-EPI — расчетная скорость клубочковой фильтрации по формуле Chronic Kidney Desease Epidemiology Collaboration; ТТГ — тиреотропный гормон; ИФР-1 — инсулиноподобный фактор роста 1. Выделенные цветом — исследования проведены в ЭНЦ.
В ходе госпитализации также подтверждено умеренное повышение уровня общего пролактина (феномен макропролактинемии исключен), рекомендованы повторное исследование сывороточной концентрации гормона с соблюдением правил сдачи анализа и магнитно-резонансная томография (МРТ) головного мозга.
С учетом множественного поражения ОЩЖ, гиперпролактинемии для исключения наследственной формы ПГПТ и определения оптимального объема хирургического лечения пациентке было проведено генетическое исследование с оценкой более 20 генов, мутации в которых ассоциированы с развитием ПГПТ. Секвенирование выполнялось на платформе Illumina NextSeq методом парно-концевого чтения (2x150 п.о.). Средняя глубина покрытия — 82x, процент целевых нуклеотидов с эффективным покрытием >10х — 99%. По результатам молекулярно-генетического тестирования в 1 экзоне гена CDKN1B (NM 004064.5) обнаружен ранее не описанный в литературе вариант (HG38, chr12:12718265 12718266insTC, c.426 427insTC) в гетерозиготном состоянии, приводящий к вставке двух нуклеотидов и сдвигу рамки считывания p.(Gly143SerfsTer3) (рис. 1).
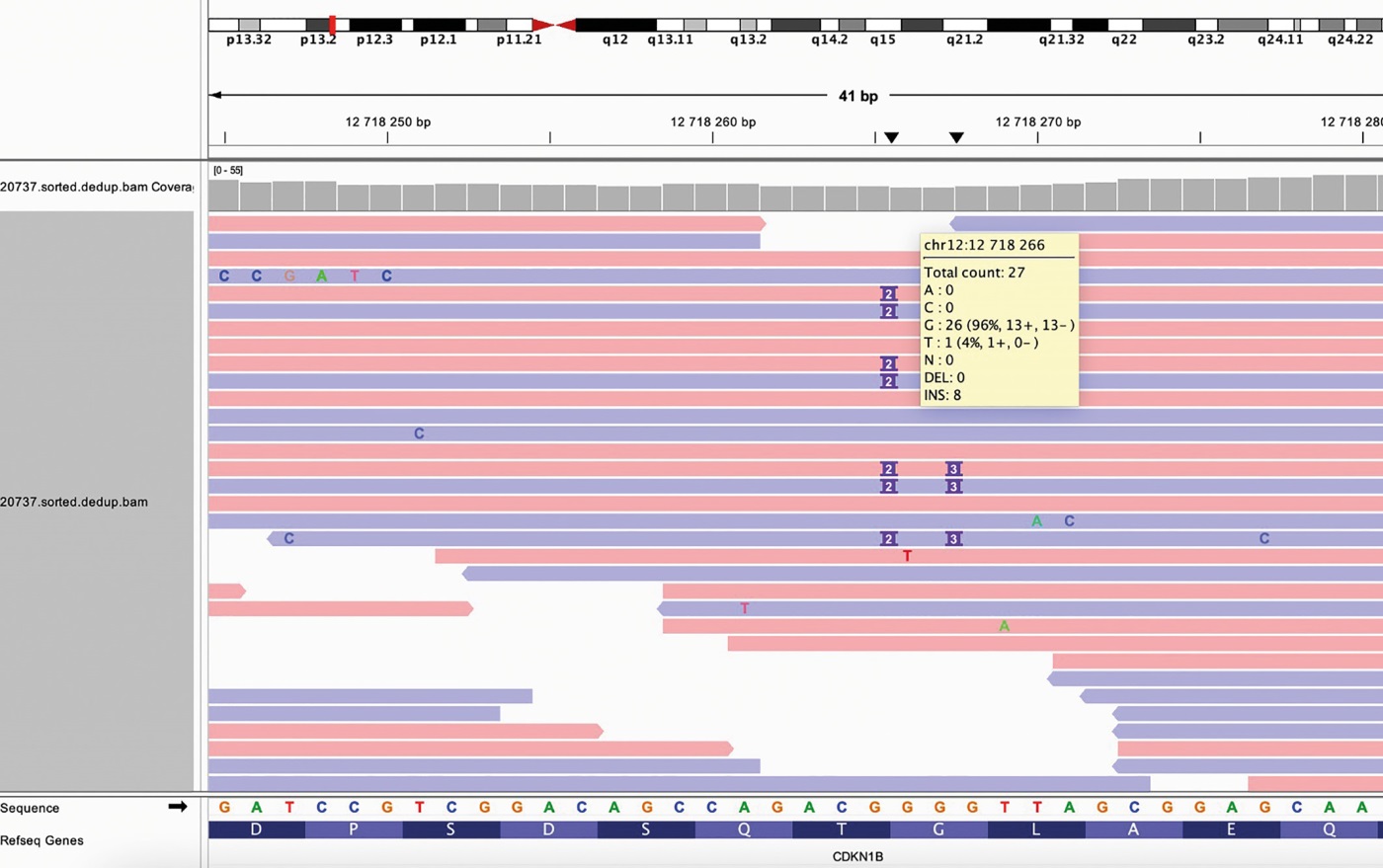
Рисунок 1. Результаты генетического исследования пациентки С.
Примечание. Массовое параллельное секвенирование (МПС, NGS) выполнено на платформе Illumina методом парно-концевого чтения (2x150 п.о.). Средняя глубина покрытия — 82x, процент целевых нуклеотидов с эффективным покрытием >10х — 99%.
В апреле 2023 г. проведено хирургическое удаление трех и резекция левой верхней ОЩЖ, при гистологическом исследовании подтверждены множественные аденомы. При интраоперационном контроле отмечено снижение сывороточной концентрации ПТГ с 70,5 пг/мл до 17,4 пг/мл через 15 минут после удаления ОЩЖ, что свидетельствовало о радикальности хирургического лечения. В послеоперационном периоде в связи с развитием гипопаратиреоза и проявлениями гипокальциемии (табл. 1) была назначена терапия карбонатом кальция 3000 мг/сут и альфакальцидолом 2 мкг/сут.
При последующем наблюдении у пациентки был диагностирован хронический гипопаратиреоз. Амбулаторно проводился динамический лабораторный контроль показателей кальций-фосфорного обмена с коррекцией медикаментозной терапии при необходимости (табл. 1).
Повторная госпитализация в ОПОЩЖ и НМО спустя 1,5 года после хирургического лечения ПГПТ. Пациентка отметила регресс суставных болей, появление судорог мышц голеней, периодического онемения пальцев рук, сохранялись общая слабость, бессонница. На фоне терапии альфакальцидолом 1 мкг утром и 1 мкг вечером, кальция карбонатом 1000 мг утром и 1000 мг вечером, магния глицинатом 200 мг на ночь была подтверждена субкомпенсация хронического послеоперационного гипопаратиреоза: выявлены целевые значения кальциемии по альбумин-скорректированному кальцию, нормофосфатемия, нормомагниемия — 0,87 ммоль/л, 25(ОН)витамин D — 103,2 нг/мл, гиперкальциурия по результатам суточного анализа мочи (табл. 1). Также из анамнеза было известно, что за месяц до поступления в стационар у пациентки произошел приступ почечной колики.
Для поиска причин субкомпенсации гипопаратиреоза в стационаре был выполнен анализ суточного профиля кальциемии (12 измерений альбумин-скорректированного кальция с интервалом забора крови в 2 часа). Учитывая верхнецелевые показатели кальциемии (рис. 2) и гиперкальциурию, были снижены дозы препаратов кальция (до 1500 мг/сут) и витамина D (до 1,5 мкг/сут).
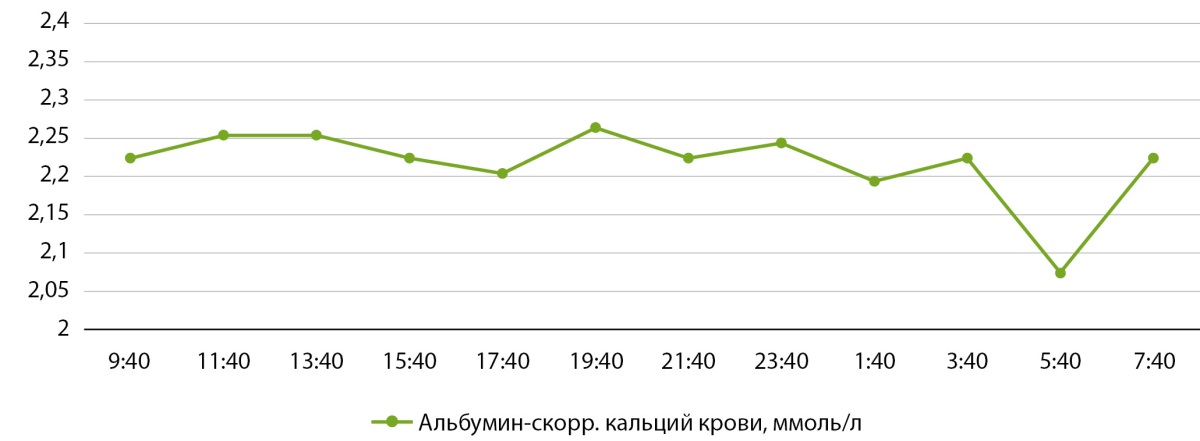
Рисунок 2. Суточный профиль кальциемии у пациентки С.
При мультиспиральной компьютерной томографии (МСКТ) определялись признаки левостороннего нефромикролитиаза, мелких кист почек (Bosniak I, II). Сывороточная концентрация маркеров костного ремоделирования соответствовала низко-нормальным значениям: остеокальцин крови — 11,1 нг/мл (15,0–46,0), щелочная фосфатаза — 62,0 Ед/л (40,0–150,0) и С-концевой телопептид коллагена I типа — 0,3 нг/мл (0,30–1,10). МПК по Т-критерию находилась в пределах нормальных значений, выявлена значимая положительная динамика в позвоночнике -0,7 SD (с 2023 г. +10,6%), в шейке -0,7 SD и бедре в целом -0,4 SD соответственно (без динамики с 2023 г.). По данным рентгенографии позвоночника в боковой проекции, признаков компрессионных переломов выявлено не было.
В рамках скрининга других потенциальных компонентов МЭН-4 были оценены функция аденогипофиза (общий пролактин — 820,8 мЕд/л (69–340), феномен макропролактинемии исключен, ИФР-1 — 204,4 нг/мл (17–238), ТТГ — 1,27 мЕд/л (0,25–3,50), кортизол в суточной моче — 242,9 нмоль/сут (100–329)) и маркеры нейроэндокринных новообразований (хромогранин А — 3,8 нмоль/л (0,0–3,0), гастрин — 90,7 пг/мл (13–115)). Таким образом, за исключением умеренной гиперпролактинемии, других значимых отклонений параметров не выявлено. При МРТ гипофиза с контрастным усилением визуализирована эндоселлярная микроаденома размером 8х5 мм. С учетом наступления менопаузы, отсутствия клинических проявлений гиперпролактинемии и малых размеров аденомы терапия агонистами дофамина не назначалась, рекомендован лабораторный и инструментальный контроль. По результатам МСКТ органов грудной клетки, брюшной полости и забрюшинного пространства с контрастным усилением диагностированы участки консолидации паренхимы легких от 3 до 14 мм в сочетании с фиброзными тяжами, не исключающие перенесенное инфекционное поражение, кальцинаты молочных желез и узелковая гиперплазия надпочечников. Пациентке был рекомендован инструментальный динамический МРТ- и МСКТ-контроль, консультация врача-пульмонолога, проведение маммографии, а также генетическое тестирование кровных родственников на предмет мутации в гене CDKN1B.
В рамках разговора о синдроме МЭН-4 нельзя не упомянуть МЭН-1, ведь изначально именно отсутствие классического генетического паттерна при наличии уже привычной клинической картины полигландулярного поражения ОЩЖ послужили отправной точкой для поиска новых возможных причин заболевания. Причиной развития синдрома МЭН 1 типа является патогенная мутация в гене — супрессоре опухолей MEN1, расположенном на хромосоме 11q13 и кодирующем менин — белок, контролирующий процессы транскрипции, деления и пролиферации клетки [3]. Классическими проявлениями синдрома МЭН-1 считаются ПГПТ (90%), НЭН ЖКТ и поджелудочной железы (30–70%), аденомы гипофиза (30–40%), реже — карциноидные опухоли, образования надпочечников, щитовидной железы, липомы, менингиомы, коллагеномы [3][4].
Заболевания, клинически соответствующие МЭН-1, но возникающие по другой причине, принято называть фенокопиями [5]. Ранее сообщалось, что около 5–25% пациентов с клиникой МЭН-1 не имеют патогенной мутации в кодирующей области гена менина [4]. Мутации в гене MEN1 обнаруживаются в среднем в 70–90% случаев семейных форм синдрома МЭН-1, в то время как при спорадических случаях эта частота значительно ниже [4]. Недавнее исследование, проведенное с участием 189 венгерских больных с типичным фенотипом MЭН-1, описало высокую распространенность фенокопий среди пациентов с неотягощенным семейным анамнезом — 74% (77/104) [6]. Причиной «невыявления» дефекта в гене MEN1 могут быть точечные мутации или делеции в некодирующих областях гена, которые не описаны на сегодняшний день [7].
Синдром МЭН-4 (OMIM #610755) впервые был описан в 2002 г. A. Fritz и соавт. как «МЭН-подобное» аутосомно-рецессивное заболевание у крыс. В течение первого года жизни у животных развивалась клиническая картина, сходная с проявлениями синдромов МЭН-1 и МЭН-2 при отсутствии мутаций в генах MEN1 и RET [8]. В 2006 г. N. Pellegata и соавт. описали мутацию гена ингибитора циклин-зависимой киназы 1В (CDKN1B) у крыс и человека с множественными поражениями эндокринных желез. Заболевание было названо синдромом МЭН-X [9]. В ходе заседания рабочей группы по изучению МЭН-синдромов в 2008 г. в г. Дельфы (Греция) МЭН-Х был переименован в МЭН-4 [10]. Среди MEN1-негативных вариантов мутации гена CDKN1B, приводящие к развитию синдрома МЭН-4, встречаются в 3% случаев [3].
Имеется две группы циклин-зависимых киназ: белки семейств p21 (CIP/KIP) и INK4 (блокируют отдельные циклин-зависимые киназы Cdk4 и Cdk6), члены которых внутри семейств обладают похожими структурными свойствами. Семейство ингибиторов p21 включает в себя три белка: p21, p27 и p57. Ген CDKN1B расположен на 12 хромосоме (12p13.1) и кодирует ингибитор циклин-зависимой киназы р27 (Kip1). Белок р27 участвует в регуляции пролиферативной активности клетки, блокируя каталитическую активность циклин-зависимых киназ D и E и, тем самым задерживая клеточный цикл в фазе G1. При мутации гена CDKN1B нарушается антимитогенная функция р27 и его возможность связываться с циклин-зависимыми киназами, регулирующими G1/S-переход [11]. Именно этот механизм лежит в основе патогенеза синдрома МЭН-4. У человека данное заболевание наследуется по аутосомно-доминантному типу, что было неоднократно подтверждено в ходе генетического обследования семей с МЭН-1-подобными проявлениями [9][2]. В отличие от белков семейства p21, ингибиторы INK4 более специфичны. В эту группу входят четыре белка: p15INK4B, p16INK4A, p18INK4C и p19INK4D. Ингибиторы семейства INK4 функционируют во время фазы G1 клеточного цикла, подавляя активность киназы CDK4, однако второй белковый продукт гена INK4A — p19ARF взаимодействует с регуляторным фактором MDM2 белка p53 и инактивирует этот фактор. Это сопровождается увеличением стабильности белка p53 и остановкой клеточного цикла [12].
Фенотипическая схожесть синдромов МЭН-1 и МЭН-4 объясняется общими путями онкогенеза. Как было сказано ранее, ингибиторы циклин-зависимой киназы служат важными регуляторами роста клеток. В исследованиях D. Franklin и соавт. было продемонстрировано, что одновременная потеря функций p27 и p18 у мышей приводит к возникновению фенотипа, сходного с синдромом МЭН-1 [13][14]. Предполагается существование эпигенетических механизмов, посредством которых менин регулирует экспрессию регуляторов пролиферации эндокринных клеток (например, путем метилирования гистона H3 Lys-4 в p27 и p18) [15]. Также в ходе исследований in vivo было показано, что изолированная потеря р27 или р18 приводила к более позднему возникновению новообразований, тогда как одновременное их выключение способствовало быстрому и агрессивному туморогенезу [13][14], что в целом может объяснять особенности МЭН-1 и МЭН-4.
Клиническое течение синдромов МЭН, а именно возраст манифестации заболевания, последовательность проявления тех или иных компонентов синдрома, их фенотипическая пенетрантность, агрессивность опухолевого роста могут служить дополнительными ориентирами для направления пациентов на генетическое исследование. Согласно имеющимся на сегодняшний день данным, для фенокопий МЭН-1 характерны более поздний срок манифестации заболевания, относительно мягкое течение и бóльшая продолжительность жизни [16].
Число наблюдений за пациентами с синдромом МЭН-4 пока весьма ограничено, а клиническое значение тех или иных вариантов мутации CDKN1B остается не до конца ясным. На 2023 г. было идентифицировано 28 различных патогенных вариантов CDKN1B [1]. Большинство из них были миссенс-мутациями (45%) и мутациями со сдвигом рамки считывания (34%), в 12% были обнаружены нонсенс-мутации, а в 9% небольшие делеции.
У нашей пациентки была обнаружена ранее не описанная в литературе мутация CDKN1B (HG38, chr12:12718265 12718266insTC, c.426 427insTC) в гетерозиготном состоянии, приводящая к вставке двух нуклеотидов и сдвигу рамки считывания p.(Gly143SerfsTer3). Манифестация заболевания пришлась на возраст 37 лет, когда впервые была выявлена гиперпролактинемия, вероятно, опухолевого генеза, с достижением ремиссии на фоне приема агонистов D2-рецепторов. Позже, в постменопаузе, диагностирован ПГПТ с вовлечением всех четырех ОЩЖ, осложненный изолированным поражением почек.
После подтверждения наследственного характера патологии проводилось обследование с целью верификации типичных проявлений МЭН-4, в ходе которого подтверждена микроаденома гипофиза (умеренное повышение пролактина до 1000 мМЕ/л в постменопаузе требует динамического наблюдения), узелковая гиперплазия обоих надпочечников. Данных за наличие НЭО ЖКТ, а также новообразований других локализаций получено не было.
Крайне малое число наблюдений за пациентами с мутациями в гене CDKN1B затрудняет определение закономерностей и характера течения этого заболевания. На 2024 г. наиболее обширное исследование с изучением корреляции генотип-фенотип у 65 пациентов с верифицированным синдромом МЭН-4 было проведено Singeisen и соавт. [1]. По их наблюдениям, МЭН-4 чаще диагностируется у женщин в постменопаузе, средний возраст манифестации — около 50 лет. Однако стоит отметить случаи очень раннего начала заболевания у 5-летней девочки с соматотропиномой [17] и 15-летнего мальчика с ПГПТ с мутацией CDKN1B [18]. Molatore и соавт. предполагают, что возраст манифестации зависит от конкретной мутации гена CDKN1B, влияющей на функции p27 [19].
Наиболее распространенным проявлением МЭН-4 (как и МЭН-1) остается ПГПТ, представленный аденомами ОЩЖ (~80% случаев), которые, в отличие от МЭН-1, лишь в 14% случаев являются множественными, а в 6% имеют рецидивирующий характер [1]. Указанные особенности ПГПТ при МЭН-4 в большинстве случаев позволяют проводить селективную паратиреоидэктомию в отличие от расширенного объема операции у пациентов с мутацией MEN1 с ревизией органов шеи. Следующим по распространенности компонентом МЭН-4 считаются аденомы гипофиза (44%). Функциональная активность образований аналогична таковым при МЭН-1: чаще всего выявляются пролактин-продуцирующие, затем сомато-, кортико- и гонадотропиномы, а также гормонально-неактивные аденомы гипофиза [20]. Микроаденомы при МЭН-4 встречаются в два раза чаще, чем макроаденомы, однако агрессивность течения этих образований и оптимальные подходы к лечению требуют дополнительной оценки. НЭО ЖКТ были выявлены лишь у 19% пациентов, в четырех из девяти случаев были обнаружены их отдаленные метастазы. ПРЩЖ описан у четырех описанных пациентов, что составило 8%. Все они были женщинами в возрасте старше 50 лет. В ¾ случаев ПРЩЖ имел мультифокальный рост, в 1 случае данные о характере роста опухоли не представлены. Информации об отдаленных результатах хирургического лечения и эффективности радиойодтерапии у этих пациенток нет. Не исключено, что наличие ПРЩЖ в данных случаях может быть связано с распространенностью заболевания в общей популяции и не быть следствием мутации в гене циклин-зависимой киназы. У трех пациентов (6,25%) с МЭН-4 были обнаружены опухоли коры надпочечников, которые были как гормонально-неактивными, так и кортизол-продуцирующими. Стоит отметить, что при синдроме МЭН-1 образования коры надпочечников имеют довольно широкую распространенность, более высокий риск злокачественного поражения и формирования АКТГ-независимой продукции кортизола по сравнению со спорадическими случаями, что требует более тщательного обследования этой когорты пациентов [21]. Эти данные нельзя однозначно экстраполировать на пациентов с МЭН-4, но, без сомнений, образования коры надпочечников при данном синдроме также требуют особой настороженности.
Синдром МЭН-4 крайне редко является наследственной причиной ПГПТ, а манифестация в среднем возрасте значимо затрудняет его диагностику, несмотря на наличие других эндокринопатий. При заболевании, ассоциированном с мутацией CDKN1B, могут формироваться как солитарные новообразования, так и полигландулярное поражение ОЩЖ. В отличие от синдрома МЭН-1, при МЭН-4-ассоциированном ПГПТ отмечено меньшее число рецидивов. Тем не менее небольшое число наблюдений за пациентами с мутациями в гене CDKN1B в настоящее время не позволяет определить закономерности течения этого заболевания, в связи с чем подробное описание клинической картины заболевания при новой выявленной мутации вносит значимый вклад в изучение данной патологии.
Источник финансирования. Статья опубликована в рамках выполнения государственного задания НИР 123021300096-3 «Новые генетические предикторы (варианты) опухолевых и неопухолевых эндокринных заболеваний у взрослых, определяемые методом полноэкзомного секвенирования, в том числе в ядерных семьях» (2023–2025 гг.).
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Все указанные авторы статьи в равной степени принимали участие в подготовке статьи согласно международным критериям авторства.
Все авторы одобрили финальную версию статьи перед публикацией, выразили согласие нести ответственность за все аспекты работы, подразумевающую надлежащее изучение и решение вопросов, связанных с точностью или добросовестностью любой части работы.
Согласие пациента. Пациент добровольно подписал информированное согласие на публикацию персональной медицинской информации в обезличенной форме.
1. Singeisen H, Melanie Renzulli M, Pavlicek V, et al. Multiple endocrine neoplasia type 4: a new member of the MEN family. Endocr Connect. 2022;12(2). doi: https://doi.org/10.1530/ec-22-0411
2. Frederiksen A, Rossing M, Hermann P, Ejersted C, Thakker R V, Frost M. Clinical Features of Multiple Endocrine Neoplasia Type 4: Novel Pathogenic Variant and Review of Published Cases. J Clin Endocrinol Metab. 2019;104(9). doi: https://doi.org/10.1210/jc.2019-00082
3. Thakker RV. Multiple endocrine neoplasia type 1 (MEN1) and type 4 (MEN4). Mol Cell Endocrinol. 2014;386(1-2). doi: https://doi.org/10.1016/j.mce.2013.08.002
4. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab. 2012;97(9). doi: https://doi.org/10.1210/jc.2012-1230
5. Мамедова Е.О., Мокрышева Н.Г., Пигарова Е.А., Пржиялковская Е.Г., Васильев Е.В., и др. Фенокопии синдрома множественных эндокринных неоплазий 1 типа: роль генов, ассоциированных с развитием аденом гипофиза // Проблемы Эндокринологии. — 2016. — Т. 62. — №4. — С. 4-10. doi: https://doi.org/10.14341/probl20166244-10
6. Kövesdi A, Tóth M, Butz H, et al. True MEN1 or phenocopy? Evidence for geno-phenotypic correlations in MEN1 syndrome. Endocrine. 2019;65(2). doi: https://doi.org/10.1007/s12020-019-01932-x
7. Мамедова Е.О., Димитрова Д.А., Белая Ж.E., Мельниченко Г.А. Роль некодирующих РНК в патогенезе синдрома множественных эндокринных неоплазий 1 типа // Проблемы Эндокринологии. — 2020. — Т. 66. — №2. — С. 4-12. doi: https://doi.org/10.14341/probl12413
8. Fritz A, Walch A, Piotrowska K, et al. Recessive transmission of a multiple endocrine neoplasia syndrome in the rat. Cancer Res. 2002;62(11)
9. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, et al. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006;103(42). doi: https://doi.org/10.1073/pnas.0603877103
10. Alevizaki M, Stratakis CA. Multiple endocrine neoplasias: advances and challenges for the future. J Intern Med. 2009;266(1):1-4. doi: https://doi.org/10.1111/j.1365-2796.2009.02108.x
11. Seabrook A, Wijewardene A, De Sousa S, et al. MEN4, the MEN1 Mimicker: A Case Series of Three Phenotypically Heterogenous Patients With Unique CDKN1B Mutations. J Clin Endocrinol Metab. 2022;107(8). doi: https://doi.org/10.1210/clinem/dgac162
12. D o i M, Hirayama J, Sassone-Corsi P. Circadian Regulator CLOCK Is a Histone Acetyltransferase. Cell. 2006;125(3). doi: https://doi.org/10.1016/j.cell.2006.03.033
13. Franklin DS, Godfrey VL, Lee H, et al. CDK inhibitors p18(INK4c) and p27(Kip1) mediate two separate pathways to collaboratively suppress pituitary tumorigenesis. Genes Dev. 1998;12(18). doi: https://doi.org/10.1101/gad.12.18.2899
14. Franklin DS, Godfrey VL, O’Brien DA, Deng C, Xiong Y. Functional Collaboration between Different Cyclin-Dependent Kinase Inhibitors Suppresses Tumor Growth with Distinct Tissue Specificity. Mol Cell Biol. 2000;20(16). doi: https://doi.org/10.1128/mcb.20.16.6147-6158.2000
15. Karnik SK, Hughes CM, Gu X, et al. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102(41). doi: https://doi.org/10.1073/pnas.0503484102
16. de Laat JM, van der Luijt RB, Pieterman CRC, et al. MEN1 redefined, a clinical comparison of mutation-positive and mutation-negative patients. BMC Med. 2016;14(1). doi: https://doi.org/10.1186/s12916-016-0708-1
17. Sambugaro S, Di Ruvo M, Ambrosio MR, et al. Early onset acromegaly associated with a novel deletion in CDKN1B 5′UTR region. Endocrine. 2015;49(1). doi: https://doi.org/10.1007/s12020-015-0540-y
18. Elston MS, Meyer-Rochow GY, Dray M, Swarbrick M, Conaglen JV. Early Onset Primary Hyperparathyroidism Associated with a Novel Germline Mutation in CDKN1B. Case Rep Endocrinol. 2015;2015. doi: https://doi.org/10.1155/2015/510985
19. Molatore S, Marinoni I, Lee M, et al. A novel germline CDKN1B mutation causing multiple endocrine tumors: Clinical, genetic and functional characterization. Hum Mutat. 2010;31(11). doi: https://doi.org/10.1002/humu.21354
20. Трухина Д.А., Мамедова Е.О., Лапшина А.М., Васильев Е.В., Тюльпаков А.Н., Белая Ж.Е. Морфологические характеристики аденом гипофиза в рамках фенокопий синдрома множественных эндокринных неоплазий 1 типа // Проблемы Эндокринологии. — 2021. — Т. 67. — №6. — С. 50-58. doi: https://doi.org/10.14341/probl12815
21. Simonds WF. Expressions of Cushing’s syndrome in multiple endocrine neoplasia type 1. Front Endocrinol (Lausanne). 2023;14. doi: https://doi.org/10.3389/fendo.2023.1183297
Петросян Альбина Сергеевна.
117036, Москва, ул. Дм. Ульянова, д.11
Researcher ID LKJ-8311-2024; Scopus Author ID 58492904500
Нет
Бибик Екатерина Евгеньевна - к.м.н.
Москва
Researcher ID AAY-3052-2020; Scopus Author ID 57195679482
Нет
Салимханов Рустам Халилович.
Москва
Scopus Author ID 57930716000
Нет
Ковалева Елена Владимировна - к.м.н.
Москва
Researcher ID T-7397-2019; Scopus Author ID 928432
Нет
Еремкина Анна Константиновна - к.м.н.
Москва
Researcher ID R-8848-2019
Нет
Уткина Марина Валерьевна - к.б.н.
Москва
Scopus Author ID 57191571945
Нет
Трошина Екатерина Анатольевна - д.м.н., профессор, член-корреспондент РАН.
Москва
Нет
Мокрышева Наталья Георгиевна - д.м.н., профессор, член-корреспондент РАН.
Москва
Нет
|
|
1. Рисунок 1. Результаты генетического исследования пациентки С. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(401KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Суточный профиль кальциемии у пациентки С. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(138KB)
|
Метаданные ▾ | |
Петросян А.С., Бибик Е.Е., Салимханов Р.Х., Ковалева Е.В., Еремкина А.К., Уткина М.В., Трошина Е.А., Мокрышева Н.Г. Новая гетерозиготная мутация в гене CDKN1B у пациентки с синдромом множественных эндокринных неоплазий 4 типа. Остеопороз и остеопатии. 2024;27(4):31-37. https://doi.org/10.14341/osteo13183
Petrosyan A.S., Bibik E.E., Salimkhanov R.H., Kovaleva E.V., Eremkina A.K., Utkina M.V., Troshina E.A., Mokrysheva N.G. A new heterozygous mutation in the CDKN1B gene in a patient affected by multiple endocrine neoplasia syndrome type 4. Osteoporosis and Bone Diseases. 2024;27(4):31-37. (In Russ.) https://doi.org/10.14341/osteo13183
117036, Москва, ул. Дмитрия Ульянова, д. 11.
Обработка персональных данных































