




Содержание
Перейти к:
https://doi.org/10.14341/osteo13178
Перейти к:
Остеомаляция — это системное заболевание скелета, характеризующееся нарушением минерализации или дефектной минерализацией вновь образованного костного матрикса у взрослых. Наиболее частой причиной является выраженный дефицит витамина D любой этиологии, дефицит кальция и дефицит фосфора (патология почек, мезенхимальные опухоли, секретирующие фактор роста фибробластов 23 (ФРФ23/FGF23), генетические заболевания). Среди генетических заболеваний чаще всего встречается X-сцепленный доминантный гипофосфатемический рахит (ген PHEX, лат. X-linked hypophosphatemic rickets (XLHR), OMIM: 307800), гораздо реже — аутосомно-доминантный гипофосфатемический рахит (ген FGF23, лат. Autosomal dominant hypophosphatemic rickets (ADHR), OMIM: 193100) и аутосомно-рецессивный гипофосфатемический рахит 1 и 2 типа (ген DMP1, ENPP1, FAM20C, лат. Autosomal recessive hypophosphatemic rickets-1 (ARHR1), OMIM: 241520; Autosomal recessive hypophosphatemic rickets-2 (ARHR2), OMIM: 613312). ADHR — крайне редкая наследственная патология, обусловленная мутацией в гене FGF23, для которой характерна манифестация в любом возрасте. В литературе описано около 50 случаев данной патологии. В статье впервые в Российской Федерации представлен клинический случай взрослой пациентки с ADHR. В ходе дифференциальной диагностики такие причины остеомаляции, как выраженный дефицит витамина D, заболевания, связанные с нарушением канальцевой реабсорбции почек, ФРФ23-продуцирующая опухоль, были исключены. Пациентке проведено генетическое исследование, по результатам которого выявлена мутация в гене FGF23, подтвержден диагноз ADHR. Инициирована терапия препаратами фосфора и активными метаболитами витамина D, на фоне которой отмечалось улучшение самочувствия в виде уменьшения болей при ходьбе, увеличения мышечной силы.
Трудности диагностики остеомаляции обусловлены отсутствием рутинного определения уровня фосфора в крови и малой осведомленностью врачей об этом заболевании. В свою очередь генетическое исследование в ряде случаев позволяет подтвердить наследственные формы, что предотвращает неоправданные хирургические вмешательства, обеспечивает своевременное назначение медикаментозной терапии и значимо улучшает качество жизни пациентов.
Сахнова Е.Е., Пржиялковская Е.Г., Мамедова Е.О., Чугунов И.С. Клинический случай аутосомно-доминантного гипофосфатемического рахита вследствие мутации в гене FGF23 у взрослого: трудности диагностики. Остеопороз и остеопатии. 2024;27(4):17-24. https://doi.org/10.14341/osteo13178
Sakhnova E.E., Przhiyalkovskaya E.G., Mamedova E.O., Chugunov I.S. A case report of autosomal dominant hypophosphatemic rickets due to a mutation in the FGF23 gene in an adult: diagnostic difficulties. Osteoporosis and Bone Diseases. 2024;27(4):17-24. (In Russ.) https://doi.org/10.14341/osteo13178
Остеомаляция — это системное заболевание скелета, характеризующееся нарушением минерализации костного матрикса у взрослых [1]. В отличие от остеопороза, развитие которого обусловлено нарушением микроархитектоники и снижением минеральной плотности костной ткани, при остеомаляции преимущественно возникает избыточное накопление неминерализованного остеоида, что приводит к размягчению костей, развитию вторичных деформаций и переломов [2]. Кальций, фосфор и щелочная фосфатаза (ЩФ) необходимы для полноценной минерализации кости, поэтому данный процесс нарушается при дефиците витамина D, снижении всасывания кальция в кишечнике, гипофосфатемии, мутациях в гене ALPL, кодирующем неспецифический тканевой изофермент ЩФ [3]. Причинами гипокальциемии и дефицита витамина D могут быть как приобретенные факторы (недостаточность инсоляции, синдром мальабсорбции, низкое содержание витамина D в пище, патология почек и печени, токсическое воздействие), так и наследственные, возникающие вследствие мутаций в генах, кодирующих 1α-гидроксилазу, 25-гидроксилазу, а также кодирующих рецептор витамина D (VDR), что приводит к развитию резистентности органов-мишеней к витамину D [4]. Причины гипофосфатемии можно разделить на 3 группы. 1. Нарушение поступления или всасывания фосфора (низкое потребление фосфатов с пищей; анорексия; парентеральное питание с недостаточным содержанием фосфатов; хроническая диарея; воспалительные заболевания кишечника; прием кальций-содержащих антацидов; прием севеламера и других лекарств, влияющих на обмен фосфора; дефицит витамина D; злоупотребление алкоголем). 2. Перераспределение фосфора внутрь клеток или в кости (острый респираторный алкалоз, декомпенсированный диабет с кетоацидозом; рефидинг синдром; синдром голодных костей; прием деносумаба). 3. Избыточные потери фосфора в почках (гиперпаратиреоз любой этиологии; патология почек (синдром Фанкони, генетические заболевания с поражение почек); ФРФ23-зависимые нарушения (ФРФ23-продуцирующая опухоль, инъекции препаратов железа; гипофосфатемический рахит другой этиологии). С точки зрения гормональной регуляции контроль обмена фосфора осуществляют паратгормон (ПТГ), ФРФ23 и 1,25(ОН)2D. Однако наиболее мощным влиянием на развитие гипофосфатемии обладает избыток фактора роста фибробластов 23 [5]. Он является фосфатурическим гормоном, который ингибирует реабсорбцию фосфатов в почках и нарушает метаболизм витамина D, в результате чего возникает гипофосфатемия, гиперфосфатурия, снижается уровень активного витамина D [6]. Среди заболеваний, ассоциированных с избытком ФРФ23 и развитием остеомаляции, наиболее актуальными являются Х-сцепленный доминантный гипофосфатемический рахит (XLHR; мутации в гене PHEX), аутосомно-доминантный гипофосфатемический рахит (ADHR; точковые миссенс-мутации в гене FGF23), синдром МакКьюна–Олбрайта–Брайцева (полиостозная фиброзная дисплазия, мутации в гене GNAS) и ФРФ23-продуцирующая опухоль. Отдельно выделяют группу редких заболеваний, приводящих к потере фосфора за счет нарушения канальцевой реабсорбции в почке без избытка ФРФ23: наследственный рецессивный гипофосфатемический рахит с гиперкальциурией (мутации в гене SLC34A3 (NaPi-IIc)); синдром Фанкони; Х-сцепленный рецессивный гипофосфатемический рахит с гиперкальциурией (болезнь Дента); отравление тяжелыми металлами [3].
Клинические симптомы остеомаляции включают в себя диффузную боль в костях, деформацию скелета (позвоночника, грудной клетки, костей таза и нижних конечностей), переломы, боли в мышцах, мышечную слабость, вплоть до атрофии [7].
При подозрении на остеомаляцию в анализе крови определяют показатели кальция, фосфора, ПТГ, ЩФ, витамин 25(ОН)D. При обнаружении гипофосфатемии проводят дифференциальную диагностику между нарушением всасывания, перераспределением и избыточным выведением фосфора. Нарушение всасывания часто сопровождается дополнительными дефицитами других макроэлементов. Перераспределение фосфора в межклеточном пространстве характерно для острых состояний, декомпенсации хронических заболеваний, что можно предположить при сборе анамнеза и наличию изменений в биохимическом анализе крови. Также исследуют индекс тубулярной реабсорбции фосфора (TRP, %), в норме он должен составлять 85–95%. Его снижение подтверждает избыточную потерю фосфора с мочой [5]. Потери фосфора c мочой могут быть обусловлены избыточным действием ФРФ23, ПТГ или первичной патологией почек. Для дифференциальной диагностики генеза остеомаляции проводят контроль электролитов в крови (натрий, калий, хлор) и рассчитывают скорость клубочковой фильтрации. Снижение данных показателей характерно для заболеваний, связанных с нарушением канальцевой реабсорбции почек.
При подтверждении избыточной потери фосфора с мочой как причины гипофосфатемии следующим оптимальным диагностическим шагом является измерение уровня ФРФ23 в крови. Однако в России, как и во многих других странах, исследование уровня ФРФ23 в рутинной практике не проводится. Его определение доступно в США, Японии, некоторых странах Европы [8]. Нормальное (относительно гипофосфатемии) или повышенное его значение наблюдается как при наследственных формах гипофосфатемических рахитов (XLHR, ADHR, ARHR1-2), так и при приобретенных состояниях (опухоль-индуцированная остеомаляция вследствие ФРФ23-процудирующих опухолей, прием некоторых форм препаратов железа (см. ниже)). При низком уровне ФРФ23 необходимо заподозрить наследственное или приобретенное первичное поражение почек (синдром Фанкони, болезнь Дента, прием антиретровирусных препаратов и пр.) [9].
Среди инструментальных исследований костная биопсия с тетрациклиновыми метками является «золотым стандартом» при дифференциальной диагностике остеомаляции, однако в клинической практике используется редко в связи с инвазивностью данной процедуры и необходимостью экспертной патоморфологической оценки. Рентгенологическими признаками заболевания являются увеличение прозрачности кости, наличие псевдопереломов (зоны трансформации Лоозера), деформация замыкательных пластинок тел позвонков в виде их вдавления («рыбьи позвонки»), низкотравматические переломы позвонков и плоских костей — ребер и грудины; наличие рахитических четок [1]. Двухэнергетическую рентгеновскую абсорбциометрию (dual-energy X-rays absorptiometry, DXA) используют для оценки минеральной плотности кости (МПК). Однако данный метод не позволяет установить причину снижения костной массы, поскольку низкие значения МПК могут наблюдаться как при остеопорозе, так и при остеомаляции. Кроме того, при некоторых формах остеомаляции (например, X-сцепленный доминатный гипофосфатемический рахит) возможны нормальные и более высокие значения МПК [10].
Для топической диагностики ФРФ23-продуцирующей опухоли проводят сцинтиграфию с радиофармпрепаратами (РФП) — 99mTc-тектротидом, 111In-октреотидом, либо позитронно-эмиссионную томографию (ПЭТ, ПЭТ/КТ) с РФП — 68Ga-DOTA-TATE. Возможно использование 18F-фтордезоксиглюкозы, однако данный РФП обладает меньшей чувствительностью в диагностике ФРФ23-продуцирующих опухолей. Для визуализации образования дополнительно используют ультразвуковое исследование (УЗИ), компьютерную томографию (КТ)/магнитно-резонансную томографию (МРТ) [3]. При наличии повышенной ЩФ для исключения болезни Педжета применяют остеосцинтиграфию с 99mTc-фосфонатами, гистологическое исследование костной ткани.
Генетическое исследование обычно проводят при манифестации признаков у молодых пациентов с целью подтверждения наследственных форм остеомаляции. Однако стоит отметить, что ADHR — крайне редкое наследственное заболевание, манифестация которого возможна и во взрослом возрасте. Поэтому при отсутствии визуализации ФРФ23-продуцирующей опухоли у немолодых пациентов также следует выполнить генетическое тестирование [11].
Дифференциальная диагностика остеомаляций необходима для определения тактики лечения: при наличии ФРФ23-продуцирующей опухоли полное излечение возможно после радикального оперативного вмешательства, в отличие от генетически обусловленных форм, коррекция которых осуществляется за счет нормализации показателей кальций-фосфорного обмена препаратами фосфора и витамина D [12].
В статье представлен клинический случай поэтапной диагностики гипофосфатемической остеомаляции, в результате которой предварительный диагноз ФРФ23-продуцирующей опухоли был исключен. После проведения генетического исследования диагностирована редкая патология — аутосомно-доминантный гипофосфатемический рахит, обусловленный мутацией в гене FGF23.
Пациентка Л. в 2018 г. в возрасте 40 лет впервые обратилась в медучреждение по месту жительства в связи с жалобами на трудность в передвижении, боли в тазобедренном, плечевом, локтевом суставах, в пояснице, мышечную слабость. Данные симптомы беспокоили с 2015 г. Также из анамнеза известно, что в раннем детстве отмечались трудности при ходьбе, был установлен диагноз «Рахит», в дальнейшем самочувствие улучшилось, физическая активность не была ограничена. Мать и отец не имеют заболеваний опорно-двигательного аппарата. Братьев, сестер и детей у пациентки нет.
В 2018 г. в ходе комплексного обследования диагностированы коксартроз, грыжи межпозвоночных дисков L4–L5, L5–S1. Консультирована неврологом, травматологом, назначена терапия нестероидными противовоспалительными средствами (нимесулид) с положительным эффектом.
В связи с ухудшением состояния, появлением сильных болей в пояснице в августе 2021 г. направлена к ревматологу по месту жительства. Установлен диагноз «Анкилозирующий спондилит», назначена терапия сульфасалазином, метотрексатом, габапентином с незначительным положительным эффектом. С февраля по октябрь 2022 г. проводилась иммунотерапия препаратом адалимумаб, без значимого эффекта.
Впервые снижение уровня фосфора в крови до 0,4 ммоль/л (0,74–1,52) выявлено в феврале 2022 г.
В октябре 2022 г. госпитализирована в ревматологическое отделение ГКБ №15 им. О.М. Филатова в г. Москве с жалобами на усиление болевого синдрома, невозможность самостоятельного передвижения без трости. При физикальном осмотре пациентки: рост — 144 см, вес — 80 кг, отмечается Х-образная деформация нижних конечностей, наличие кифоза в грудном отделе позвоночника. По результатам лабораторного обследования повторно подтверждена гипофосфатемия (фосфор — 0,68 ммоль/л (0,8–1,45)), выявлена гиперфосфатурия (фосфор в моче — 43,2 ммоль/л (12–42)), снижение кальция, скорректированного на альбумин до 2,11 ммоль/л (2,15–2,55) при нормальном уровне ПТГ. В ходе инструментальной диагностики по данным КТ тазобедренных суставов и костей таза отмечались участки разряжения костной ткани в правой подвздошной кости (в области крестцово-подвздошного сочленения) и теле позвонка L4. Консультирована эндокринологом, учитывая характерную клиническую картину, заподозрена фосфопеническая остеомаляция, рекомендована терапия альфакальцидолом.
Впервые госпитализирована в ГНЦ РФ ФБГУ «НМИЦ эндокринологии» Минздрава России в январе 2023 г. Проведено лабораторное обследование, по результатам которого (на фоне терапии альфакальцидолом, 3 мкг/сут) подтверждена гипофосфатемия до 0,4 ммоль/л (0,74–1,52), выявлено снижение индекса тубулярной реабсорбции фосфора до 53% (при норме 85–95%), недостаточность 25(ОН)D (26 нг/мл), низко-нормальный уровень кальция, скорректированного на альбумин; при этом показатели ПТГ и ЩФ в крови — в пределах референсных значений.
Для исключения заболеваний, связанных с нарушением канальцевой реабсорбции почек, дополнительно исследованы электролиты и уровень креатинина в крови, по результатам: показатели натрия, калия в норме, снижения скорости клубочковой фильтрации (СКФ) не выявлено.
В ходе инструментального обследования на серии постуральных рентгенограмм нижних конечностей в режиме лазерной склейки определяется умеренная Х-образная деформация с периостальными утолщениями, умеренный гипертрофический остеопороз, двухсторонний коксартроз 2 степени, артроз крестцово-подвздошных сочленений 2 степени, укорочение левой конечности на 9,44 мм, двухсторонний гонартроз 1–2 степени, двухсторонний артроз голеностопных суставов 1–2 степени, ротационные подвывихи в суставах (рис. 1). По результатам КТ, визуализированы участки разрежения костной ткани в Th10, L1, L4, перелом заднего отрезка 10 ребра и перелом тела правой подвздошной кости. По данным DXA, отмечалось снижение МПК в проксимальном отделе бедренной кости: neck до -2,3SD по Z-критерию, total до -5,0SD по Z-критерию; МПК в поясничном отделе позвоночника соответствовала возрастной норме: L1–L4 -0,2SD по Z-критерию.
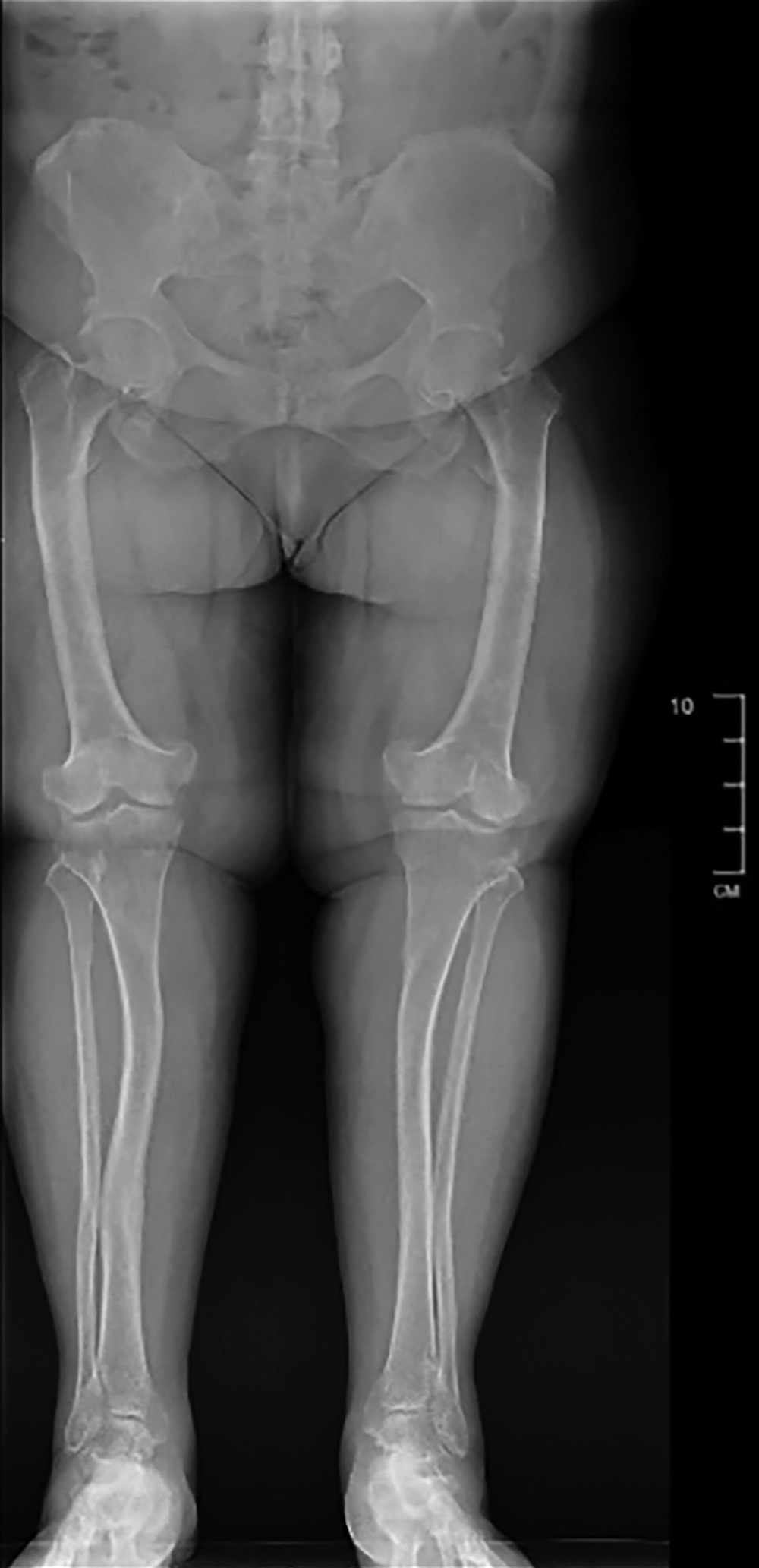
Рисунок 1. Рентгенологическое исследование нижних конечностей в режиме лазерной склейки.
Таким образом, учитывая возраст пациентки, данные лабораторного и инструментального обследования, заподозрена фосфопеническая остеомаляция опухолевого генеза (ФРФ23-продуцирующая опухоль). В ходе топической диагностики по результатам МРТ грудного и поясничного отделов позвоночника, крестцово-подвздошных сочленений, КТ с контрастированием органов грудной клетки, брюшной полости, малого таза, опухоль не визуализирована. По данным МРТ головного мозга выявлено образование лобной кости справа размерами 5,9х3,2х5 мм. Для подтверждения наличия ФРФ23-продуцирующей опухоли рекомендована сцинтиграфия с 99mTc-тектротидом.
За время госпитализации скорректирована доза альфакальцидола до 4 мкг/сут, к терапии добавлены препараты кальция (кальция карбонат 500 мг 3 раза в сутки), фосфора (калия дигидрофосфат, натрия двухосновной фосфора 450 мг по 1 капсуле 4 раза в день) и нативной формы витамина D в насыщающей дозе на 1 месяц с последующим переходом на поддерживающую дозу. На фоне проводимой терапии пациентка отмечала улучшение самочувствия в виде уменьшения болевого синдрома, увеличения мышечной силы.
После выписки проведена сцинтиграфия с 99mTc-тектротидом, по результатам которой очагов патологической гиперфиксации 99mTc-тектротида не обнаружено, ОФЭКТ-КТ признаков гормонально-активных образований с гиперэкспрессией соматостатиновых рецепторов не выявлено.
Через 8 месяцев пациентка госпитализирована повторно в ГНЦ РФ ФБГУ «НМИЦ эндокринологии» Минздрава России. В ходе лабораторного обследования (на фоне терапии альфакальцидолом 2 мкг 2 раза в сутки, колекальциферолом 2500 МЕ (5 капель) в сутки, калия дигидрофосфатом 450 мг 4 раза в сутки, кальция карбонатом 500 мг 3 раза в сутки) пациентка отметила улучшение самочувствия: расширение физической нагрузки, уменьшение болевого синдрома. При лабораторном исследовании наблюдалась положительная динамика в виде повышения уровня фосфора до 0,6 ммоль/л, нормализации кальция, скорректированного на альбумин (в таблице 1 представлены данные динамического лабораторного обследования в ГНЦ РФ ФБГУ «НМИЦ эндокринологии» Минздрава России). По данным DXA отмечалось значимое увеличение показателей минеральной плотности костной ткани в проксимальном отделе бедренной кости: neck до 0,2 SD по Z-критерию, total до -0,1 SD по Z-критерию.
Таблица 1. Результаты динамического лабораторного обследования пациентки Л. в ходе госпитализаций в ГНЦ РФ ФБГУ «НМИЦ эндокринологии» Минздрава России
|
Показатель |
Референсные значения |
18.01.2023 |
10.08.2023 |
22.06.2024 |
|
Креатинин, мкмоль/л |
50–98 |
48 |
56,8 |
59,5 |
|
Са, скорр. на альбумин, ммоль/л |
2,15–2,55 |
2,17 |
2,19 |
2,13 |
|
Фосфор, ммоль/л |
0,74–1,52 |
0,41 |
0,6 |
0,4 |
|
Паратгормон, пг/мл |
15–65 |
58,3 |
64 |
64 |
|
Щелочная фосфатаза, Ед/л |
40–150 |
122 |
149 |
130 |
|
25 (OH) Витамин D (Liaison), нг/мл |
30–100 |
26,2 |
- |
24 |
|
Индекс тубулярной реабсорбции фосфора, % |
85–95 |
53 |
52,7 |
44,9 |
|
Кальций в суточной моче, ммоль/сут |
2,5–8 |
- |
4,82 |
5,63 |
|
Гемоглобин, г/л |
112–153 |
142 |
- |
138 |
|
Ферритин, нг/мл |
15–160 |
- |
- |
32,8 |
Для оценки безопасности терапии проведено исследование кальция в суточной моче (показатель в норме) и КТ органов брюшной полости, по результатам которого микролиты в почках не визуализировались.
Учитывая отсутствие убедительных данных за наличие опухоли, рахит в анамнезе, для уточнения наследственного генеза гипофосфатемии проведено генетическое исследование — высокопроизводительное параллельное секвенирование панели генов-кандидатов, ассоциированных с нарушениями фосфорно-кальциевого обмена (NextSeq550, Illumina, США), по результатам которого в гене FGF23 в экзоне 3 обнаружен патогенный вариант (HG38, chr12:4370563C>T, c.536G>A) в гетерозиготном состоянии, приводящий к аминокислотной замене p.Arg179Gln, что подтверждает наличие аутосомно-доминантного гипофосфатемического рахита.
В ходе последующей госпитализации летом 2024 г. с целью динамического обследования проведена оценка показателей фосфорно-кальциевого обмена (табл. 1), по результатам которой скорректирована доза альфакальцидола до 2 мкг/сут, возобновлен прием колекальциферола в насыщающей дозе. По данным DXA наблюдалась положительная динамика: увеличение МПК в шейке бедренной кости до 0,4SD по Z-критерию, в поясничном отделе позвоночника (L1–L4) до 1,8 SD по Z-критерию. Клинически пациентка отмечает улучшение самочувствия, расширение физической активности (уменьшение болевого синдрома при ходьбе) на фоне проводимой терапии.
Аутосомно-доминантный гипофосфатемический рахит (ADHR) представляет собой крайне редкое наследственное заболевание, вызванное гетерозиготными точковыми мутациями в гене FGF23, который активно участвует в гомеостазе фосфора [13]. В результате транскрипции гена FGF23, расположенного на 12-и хромосоме и состоящего из 3 экзонов, синтезируется белок ФРФ23, состоящий из 251 аминокислоты (32 kD) [14]. Он имеет уникальную структуру и содержит сайт протеолитического расщепления с определенной последовательностью аминокислот со 176-й по 179-ю: Arg176-His177-Thr178-Arg179. За счет протеолиза происходит инактивация ФРФ23. Мутации, локализованные в одном из двух аргининов, такие как p.Arg176Gln, p.Arg179Gln, p.Arg176Trp и p.Arg179Trp, нарушают ферментативное расщепление белка ФРФ23 фуриноподобной протеинконвертазой. Это приводит к сохранению активной формы белка, что, в свою очередь, обуславливает увеличение экскреции фосфатов с мочой [15].
В литературе описано около 50 клинических случаев ADHR, при этом у большинства пациентов диагноз был установлен уже во взрослом возрасте [16]. В российской литературе нами найдено описание лишь одного клинического случая ADHR у ребенка [17].
Клиническая картина данного заболевания неоднородна и зависит от возраста пациента на момент начала заболевания и степени тяжести гипофосфатемии. У детей она характеризуется признаками рахита: деформации конечностей (вальгус/варус), боли в костях, непропорциональность и снижение темпов роста, деформации челюсти. Если манифестация возникает в более зрелом возрасте, то клинические симптомы включают боль в костях, мышечную слабость и псевдопереломы. У части пациентов заболевание протекает бессимптомно на протяжении всей жизни, у некоторых чередуются периоды наличия и отсутствия симптомов [16]. Именно вариант волнообразного течения наблюдается у пациентки Л. В детстве у нее отмечались деформации конечностей, непропорциональность роста, в связи с чем был диагностирован рахит. В течение последующих 25–30 лет симптомы остеомаляции пациентку не беспокоили, физическая активность не была ограничена. Только в возрасте 37 лет возникла повторная манифестация в виде выраженного болевого синдрома в костях и суставах, трудности при ходьбе, мышечной слабости. Эту особенность объясняют различной степенью устойчивости белка ФРФ23 к расщеплению в зависимости от конкретного типа мутации. Некоторые исследования in vitro показали, что мутация в положении 179 обуславливает синтез более устойчивого к расщеплению белка ФРФ23 по сравнению с тем, что образуется вследствие мутации в положении 176 [18].
ADHR в первую очередь необходимо дифференцировать с онкогенной остеомаляцией, а также с другими наследственными формами рахита. К лабораторным признакам заболевания относят гипофосфатемию в сочетании с низким уровнем индекса тубулярной реабсорбции фосфатов (менее 85%). Другие лабораторные изменения неспецифичны и включают в себя гиперфосфатурию, нормальный уровень кальция или его снижение при нормальном или повышенном уровне ПТГ, отмечается снижение 1,25(ОН)2D при нормальном уровне 25(OH)D [3]. Для подтверждения избытка ФРФ23 возможно определение его уровня в крови, однако данный анализ в клинической практике в России не зарегистрирован.
В связи с отсутствием рутинного измерения уровня фосфора в биохимическом анализе крови, малой осведомленностью врачей о данном заболевании своевременная постановка диагноза фосфопенической остеомаляции представляет собой сложную задачу. У пациентки Л. только спустя 7 лет от момента возобновления симптомов впервые исследован уровень фосфора в крови и выявлена гипофосфатемия. В ходе дальнейшего обследования подтверждено снижение индекса тубулярной реабсорбции фосфатов, снижение кальция крови при нормальном уровне паратгормона и ЩФ. По результатам инструментальной диагностики, визуализированы участки разрежения костной ткани, перелом ребра и подвздошной кости, отмечено снижение МПК. В связи с выявленными изменениями заподозрена фосфопеническая остеомаляция.
При возникновении заболевания во взрослом возрасте в первую очередь проводится топическая диагностика для исключения гипофосфатемической остеомаляции опухолевого генеза [19]. У пациентки Л., по данным МРТ головного мозга, обнаружено образование лобной кости, однако по результатам более чувствительного метода — сцинтиграфии с 99mTc–тектротидом — гормонально-активных образований не выявлено. Таким образом, учитывая особенности анамнеза, для уточнения причины остеомаляции было проведено генетическое исследование, по результатам которого подтвержден диагноз аутосомно-доминантного гипофосфатемического рахита.
В связи с небольшой распространенностью ADHR в литературе ограничены сведения по ведению данных пациентов. Терапевтические подходы основаны на рекомендациях по лечению пациентов с XLH — самой частой формой гипофосфатемических рахитов, обусловленной мутацией в гене PHEX. Терапией первой линии являются препараты фосфора и активной формы витамина D. Пероральные фосфаты назначаются в дозе около 15 до 60 мг/кг массы тела в сутки (средняя суточная доза 1–3 г) в 3–5 приемов из-за короткого периода полувыведения (около 4 часов). Дозу активного аналога витамина D (альфакальцидол/кальцитриол) следует титровать в зависимости от возраста 15–60 нг/кг/сут, (суточная доза для альфакальцидола — 1–4 мкг, для кальцитриола — 0,25–2 мкг) в 2–3 приема [20]. Дополнительно могут быть назначены нативные препараты витамина D (колекальциферол) для компенсации недостаточности 25(ОН)D. Существуют исследования, показывающие, что высокие концентрации альфакальцидола могут стимулировать синтез ФРФ23 и усугублять гипофосфатемию, что важно учитывать при назначении терапии [21].
Эффективность лечения оценивают клинически по улучшению общего состояния, уменьшению болевого синдрома и мышечной слабости, а также по уровню показателей кальций-фосфорного обмена. У пациентки Л. при приеме альфакальцидола 4 мкг/сут не удалось достичь значимого повышения уровня фосфора крови, в связи с чем дополнительно были рекомендованы препараты фосфора. На этом фоне пациентка отмечала улучшение самочувствия в виде уменьшения болевого синдрома, увеличения мышечной силы.
Однако необходимо помнить о побочных эффектах данной терапии в виде развития нефрокальциноза (вследствие передозировки метаболитами витамина D) и вторичного гиперпаратиреоза (обусловленного развитием гипокальциемии на фоне приема фосфатов). Это требует тщательного мониторинга показателей кальций-фосфорного обмена в крови, суточной кальцийурии с последующей коррекцией дозы препаратов при необходимости [22][23].
У пациентки Л. на фоне проводимой терапии кальций в суточной моче — в пределах референсных значений, по результатам КТ — микролиты в почках не обнаружены.
Интересен вопрос взаимосвязи гомеостаза фосфора и железа. Имеются данные, что экспрессия FGF23 увеличивается при дефиците железа, а в сочетании с нарушением расщепления ФРФ23 при ADHR это приводит к более высоким концентрациям интактного ФРФ23 и выраженной гипофосфатемии. В связи с чем у части пациентов с ADHR пероральная терапия препаратами железа способствовала нормализации уровня ФРФ23, фосфора крови и улучшала прогноз [24]. Также примечательно, что все пациенты с отсроченным дебютом ADHR — женщины репродуктивного возраста, и манифестация заболевания была ассоциирована с пубертатом, беременностью и родами, то есть состояниями, ассоциированными с дефицитом железа [25]. Однако стоит отметить, что, по данным литературы, у пациентов с анемией, но без аутосомно-доминантного гипофосфатемического рахита восполнение дефицита железа путем введения некоторых парентеральных препаратов (в частности железа карбоксимальтозат и реже — железо-сахарозный комплекс) вызывает резкое повышение интактного ФРФ23, что, в свою очередь, приводит к тяжелой гипофосфатемии. Поэтому у пациентов с ADHR внутривенная инфузия вышеуказанных препаратов может спровоцировать еще более значимое ухудшение состояния. Механизм данного эффекта неясен, но, по-видимому, напрямую связан с углеводородной частью комплекса железа, которая влияет на степень расщепления ФРФ23 [26][27]. Таким образом, для коррекции дефицита железа у пациентов с ADHR рекомендовано использовать именно таблетированные препараты.
Еще одним потенциальным вариантом лечения пациентов с ADHR является буросумаб. Он представляет собой моноклональное антитело, которое связывает избыток ФРФ23, ингибирует его, тем самым снижая гиперфосфатурию. В 2018 г. препарат одобрен FDA для лечения детей с Х-сцепленным гипофосфатемическим рахитом [28]. По данным исследований, буросумаб более эффективен, чем стандартная терапия препаратами фосфора и витамина D. Снижая экскрецию фосфора с мочой, препарат нормализует уровень фосфора в крови и обеспечивает клинически значимые улучшения в виде снижения тяжести рахита у пациентов с Х-сцепленной гипофосфатемией [29]. Однако до сих пор нет исследований по применению буросумаба у пациентов с ADHR, что ограничивает его применение.
После установления стабильной дозы препаратов (фосфатов, витамина D, буросумаба) рекомендовано проводить динамическое обследование с оценкой уровня сывороточного фосфора, кальция, креатинина и щелочной фосфатазы, а также кальция в суточной моче примерно каждые 3–4 месяца, уровень ПТГ — 1 раз в 6 месяцев и при наличии гиперкальциемии. Важно отметить, что сохранение гипофосфатемиии натощак само по себе не является показанием для корректировки дозы препаратов, так как при оценке эффективности лечения необходимо учитывать не только лабораторные показатели, но и клиническую картину в виде улучшения общего самочувствия, уменьшения тяжести симптомов заболевания [6].
Остеомаляция так же, как и остеопороз, имеет большое социально-экономическое значение в связи с повышением риска возникновения низкотравматических переломов и инвалидизации пациентов.
Своевременная диагностика важна для определения дальнейшей тактики ведения пациента, однако представляет трудности в связи с неспецифическим характером клинических симптомов, отсутствием рутинного определения уровня фосфора в крови и малой осведомленностью врачей об этом заболевании. В ряде случаев генетическое исследование необходимо для дифференциальной диагностики и исключения наследственных форм остеомаляции. Это позволяет своевременно назначить медикаментозную терапию и значимо улучшить качество жизни пациентов.
Источники финансирования. Работа выполнена в рамках государственного задания «Новые технологии диагностики и дифференциальной диагностики первичного и вторичного остеопороза на фоне эндокринопатий и орфанных заболеваний скелета» № 124020700097-8.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Участие авторов. Все авторы внесли значимый вклад в написание статьи, прочли и одобрили финальный вариант рукописи до публикации.
Согласие пациента. Пациент добровольно подписал информированное согласие на публикацию персональной медицинской информации в обезличенной форме.
1. Fukumoto S, Ozono K, Michigami T, et al. Pathogenesis and diagnostic criteria for rickets and osteomalacia—proposal by an expert panel supported by the Ministry of Health, Labour and Welfare, Japan, the Japanese Society for Bone and Mineral Research, and the Japan Endocrine Society. J Bone Miner Metab. 2015;33(5):467-473. doi: https://doi.org/10.1007/s00774-015-0698-7
2. Vieth R. Weaker bones and white skin as adaptions to improve anthropological “fitness” for northern environments. Osteoporos Int. 2020;31(4):617-624. doi: https://doi.org/10.1007/s00198-019-05167-4
3. Голоунина О.О., Рунова Г.Е., Фадеев В.В. Остеомаляция в практике эндокринолога: этиология, патогенез, дифференциальная диагностика с остеопорозом // Остеопороз и остеопатии. — 2019. — Т. 22. — №2. — С. 23-31. doi: https://doi.org/10.14341/osteo12117
4. Biasucci G, Donini V, Cannalire G. Rickets Types and Treatment with Vitamin D and Analogues. Nutrients. 2024;16(3):1-15. doi: https://doi.org/10.3390/nu16030416
5. Гронская С.А., Белая Ж.Е., Мельниченко Г.А. ФРФ23-индуцированная остеомаляция опухолевого генеза // Проблемы эндокринологии. — 2022. — Т. 68. — №5. — С. 56-66. doi: https://doi.org/10.14341/probl13130
6. Ackah SA, Imel EA. Approach to Hypophosphatemic Rickets. J Clin Endocrinol Metab. 2023;108(1):209-220. doi: https://doi.org/10.1210/clinem/dgac488
7. Gifre L, Peris P, Monegal, A. et al. Osteomalacia revisited: A report on 28 cases. Clin Rheumatol. 2011;30(5):639-645. doi: https://doi.org/10.1007/s10067-010-1587-z
8. Heijboer AC, Cavalier E. The Measurement and Interpretation of Fibroblast Growth Factor 23 (FGF23) Concentrations. Calcif Tissue Int. 2023;112(2):258-270. doi: https://doi.org/10.1007/s00223-022-00987-9
9. Florenzano P, Cipriani C, Roszko KL, et al. Review Approach to patients with hypophosphataemia. LANCET Diabetes Endocrinol. 2020;8587(19). doi: https://doi.org/10.1016/S2213-8587(19)30426-7
10. Rosenthall L. DEXA bone densitometry measurements in adults with X-linked hypophosphatemia. Clin Nucl Med. 1993;18(7):564-566. doi: https://doi.org/10.1097/00003072-199307000-00004
11. Von Falck C, Rodt T, Rosenthal H, et al. 68Ga-DOTANOC PET/CT for the detection of a mesenchymal tumor causing oncogenic osteomalacia. Eur J Nucl Med Mol Imaging. 2008;35(5):1034. doi: https://doi.org/10.1007/s00259-008-0755-8
12. Cianferotti L. Osteomalacia Is Not a Single Disease. Int J Mol Sci. 2022;23(23). doi: https://doi.org/10.3390/ijms232314896
13. White KE, Carn G, Lorenz-Depiereux B, et al. Autosomal-dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF-23. Kidney Int. 2001;60(6):2079-2086. doi: https://doi.org/10.1046/j.1523-1755.2001.00064.x
14. Hui Q, Jin Z, Li X, et al. FGF family: From drug development to clinical application. Int J Mol Sci. 2018;19(7). doi: https://doi.org/10.3390/ijms19071875
15. Li X. The FGF metabolic axis. Front Med. 2019;13(5):511-530. doi: https://doi.org/10.1007/s11684-019-0711-y
16. Mameli C, Sangiorgio A, Colombo V, et al. Autosomal dominant hypophosphatemic rickets: A case report and review of the literature. Int J Environ Res Public Health. 2021;18(16):4-13. doi: https://doi.org/10.3390/ijerph18168771
17. Куликова К.С., Васильев Е.В., Петров В.М., Тюльпаков А.Н. Аутосомно-доминантный гипофосфатемический рахит, обусловленный мутацией в гене FGF23, – первый клинический случай в России // World Journal of Personalized Medicine. — 2018. — Т. 2. — №1. — С 5-9. doi: https://doi.org/10.14341/pm9661
18. Liu C, Zhao Z, Wang O, et al. Earlier Onset in Autosomal Dominant Hypophosphatemic Rickets of R179 than R176 Mutations in Fibroblast Growth Factor 23: Report of 20 Chinese Cases and Review of the Literature. Calcif Tissue Int. 2019;105(5):476-486. doi: https://doi.org/10.1007/s00223-019-00597-y
19. Гронская С.А., Белая Ж.Е., Рожинская Л.Я., и др.. Клинические проявления, принципы диагностики и лечения ФРФ23-секретирующих опухолей: результаты наблюдения 40 случаев // Проблемы эндокринологии. — 2023. — Т. 69. — №5. — С. 25-38. doi: https://doi.org/https://doi.org/10.14341/probl13221
20. Linglart A, Biosse-Duplan M, Briot K, et al. Therapeutic management of hypophosphatemic rickets from infancy to adulthood. Endocr Connect. 2014;3(1):R13-R30. doi: https://doi.org/10.1530/ec-13-0103
21. Zittermann A, Berthold HK, Pilz S. The effect of vitamin D on fibroblast growth factor 23: a systematic review and meta-analysis of randomized controlled trials. Eur J Clin Nutr. 2021;75(6):980-987. doi: https://doi.org/10.1038/s41430-020-00725-0
22. Tieder M, Blonder J, Strauss S, et al. Hyperoxaluria is not a cause of nephrocalcinosis in phosphate-treated patients with hereditary hypophosphatemic rickets. Nephron. 1993;64(4):526-531. doi: https://doi.org/10.1159/000187395
23. Carpenter TO, Mitnick MA, Ellison A, et al. Nocturnal hyperparathyroidism: A frequent feature of X-Linked hypophosphatemia. J Clin Endocrinol Metab. 1994;78(6):2-7. doi: https://doi.org/10.1210/jcem.78.6.8200940
24. Imel EA, Liu Z, Coffman M, et al. Oral Iron Replacement Normalizes Fibroblast Growth Factor 23 in Iron-Deficient Patients With Autosomal Dominant Hypophosphatemic Rickets. J Bone Miner Res. 2020;35(2):231-238. doi: https://doi.org/10.1002/jbmr.3878
25. Imel EA, Hui SL, Econs MJ. FGF23 Concentrations Vary With Disease Status in Autosomal Dominant Hypophosphatemic Rickets. 2007;22(4):520-526. doi: https://doi.org/10.1359/JBMR.070107
26. Klein K, Asaad S, Econs M, Rubin JE. Severe FGF23-based hypophosphataemic osteomalacia due to ferric carboxymaltose administration. BMJ Case Rep. 2018;2018. doi: https://doi.org/10.1136/bcr-2017-222851
27. Shimizu Y, Tada Y, Yamauchi M, et al. Hypophosphatemia induced by intravenous administration of saccharated ferric oxide. Another form of FGF23-related hypophosphatemia. Bone. 2009;45(4):814-816. doi: https://doi.org/10.1016/j.bone.2009.06.017
28. Carpenter TO, Whyte MP, Imel EA, et al. Burosumab Therapy in Children with X-Linked Hypophosphatemia. N Engl J Med. 2018;378(21):1987-1998. doi: https://doi.org/10.1056/nejmoa1714641
29. Imel EA, Glorieux FH, Whyte MP, et al. Burosumab versus conventional therapy in children with X-linked hypophosphataemia: a randomised, active-controlled, open-label, phase 3 trial. Lancet. 2019;393(10189):2416-2427. doi: https://doi.org/10.1016/S0140-6736(19)30654-3
Сахнова Екатерина Евгеньевна.
111539, Москва, ул. Вешняковская, д. 23
Нет
Пржиялковская Елена Георгиевна - к.м.н.
Москва
Нет
Мамедова Елизавета Октаевна - к.м.н.
Москва
Нет
Чугунов Игорь Сергеевич - к.м.н.
Москва
Нет
|
|
1. Рисунок 1. Рентгенологическое исследование нижних конечностей в режиме лазерной склейки. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(151KB)
|
Метаданные ▾ | |
Сахнова Е.Е., Пржиялковская Е.Г., Мамедова Е.О., Чугунов И.С. Клинический случай аутосомно-доминантного гипофосфатемического рахита вследствие мутации в гене FGF23 у взрослого: трудности диагностики. Остеопороз и остеопатии. 2024;27(4):17-24. https://doi.org/10.14341/osteo13178
Sakhnova E.E., Przhiyalkovskaya E.G., Mamedova E.O., Chugunov I.S. A case report of autosomal dominant hypophosphatemic rickets due to a mutation in the FGF23 gene in an adult: diagnostic difficulties. Osteoporosis and Bone Diseases. 2024;27(4):17-24. (In Russ.) https://doi.org/10.14341/osteo13178
117036, Москва, ул. Дмитрия Ульянова, д. 11.
Обработка персональных данных































