



Содержание
Перейти к:
 А. Л. Кунгурцева,
Ю. В. Тихонович,
И. Ю. Кульгускин,
Н. А. Шолохова,
Ю. В. Максимова,
А. В. Витебская
А. Л. Кунгурцева,
Ю. В. Тихонович,
И. Ю. Кульгускин,
Н. А. Шолохова,
Ю. В. Максимова,
А. В. Витебская https://doi.org/10.14341/osteo13204
Перейти к:
Синдромы преждевременного старения — это группа ультраредких гетерогенных наследственных заболеваний, проявляющихся преимущественно в детском возрасте и характеризующихся ускоренным старением организма. Несмотря на различия патогенеза, заболевания характеризуются мультисистемными изменениями, в том числе поражением опорно-двигательного аппарата, которые представлены множественными контрактурами суставов, деформациями позвоночника и конечностей, изменениями структуры костной ткани.
Проанализированы данные обследования 6 пациентов: 5 детей (3 мальчика и 2 девочки) с детской прогерией (синдром Хатчинсона-Гилфорда) (4 пациента с классическим генотипом детской прогерии (c.1824C>T в гене LMNA) и 1 ребенок — с неклассическим (c.1968+1G>A в гене LMNA)) и одна девочка с неонатальным прогероидным синдромом (синдром Видемана-Раутенштрауха) (c.3337- 11T>C/ c.3677T>C в гене POLR3A). Диагноз был установлен в возрасте 1,9 (1,5; 4,3) года (Ме (25%; 75%)). Пациенты находились под наблюдением врача — детского эндокринолога, осматривались врачом — травматологом-ортопедом, проводились рентгенологические исследования, денситометрия поясничного отдела позвоночника. Возраст на момент первичного обследования — 6,0 (3,5; 7,2) лет, повторного — 7,6 (7,5; 9,3) года.
Костно-суставные изменения при синдроме Хатчинсона-Гилфорда были представлены контрактурами межфаланговых суставов пальцев кистей и стоп, лучезапястных, локтевых, тазобедренных, коленных и голеностопных суставов, плоско-вальгусными стопами; у пациента с неклассическим генотипом детской прогерии данные изменения диагностированы при первом обследовании в 1 год 6 мес, что подтверждает тяжелое течение заболевания при данном генотипе. У двух пациентов старшего возраста (7 лет 5 мес и 9 лет 10 мес) также диагностированы coxa valga с двух сторон с развитием асептического некроза головки бедренной кости и закрытого вывиха левой бедренной кости. При неонатальном прогероидном синдроме поражения опорно-двигательного аппарата проявлялись в виде множественных контрактур больших и малых суставов и деформацией позвоночника. Костный возраст либо соответствовал хронологическому, либо отставал на 18 (15; 26) мес. У всех пациентов, по данным денситометрии, был диагностирован остеопороз (Z-критерий: -3,4 (-3,0; -3,8)); переломов не зафиксировано.
Выявленные изменения костной ткани и опорно-двигательного аппарата у наших пациентов соответствуют особенностям, описанным в мировой литературе у пациентов с синдромами Хатчинсона-Гилфорда и Видемана-Раутенштрауха. Схожесть патологических изменений свидетельствует о схожести фенотипов заболеваний, входящих в группу синдромов преждевременного старения.
Кунгурцева А.Л., Тихонович Ю.В., Кульгускин И.Ю., Шолохова Н.А., Максимова Ю.В., Витебская А.В. Особенности опорно-двигательного аппарата и костной ткани у пациентов с синдромами преждевременного старения. Остеопороз и остеопатии. 2025;28(3):34-45. https://doi.org/10.14341/osteo13204
Kungurtseva A.L., Tikhonovich Y.V., Kulguskin I.Y., Sholokhova N.A., Maksimova J.V., Vitebskaya A.V. Features of musculoskeletal apparatus and bone tissue in patients with premature aging syndromes. Osteoporosis and Bone Diseases. 2025;28(3):34-45. (In Russ.) https://doi.org/10.14341/osteo13204
Прогероидные синдромы (или синдромы преждевременного старения) — это группа ультраредких наследственных заболеваний, характеризующихся ускоренным старением организма с поражением большинства органов и систем, в том числе изменениями опорно-двигательного аппарата [1–2].
Чаще всего в литературе встречается описание детской прогерии (синдром Хатчинсона-Гилфорда, распространенность 1:20 000 000), для которой типичен специфичный фенотип: низкий рост с деформациями конечностей, тяжелый дефицит массы тела, генерализованная липодистрофия, тонкая морщинистая гиперпигментированная кожа с выступающим венозным рисунком, микрогнатия, узкий клювовидный нос, тонкие губы, маленький рот, зубочелюстные и костно-суставные аномалии. Такая форма, как правило, манифестирует примерно с 9–18 месяцев жизни [1][3].
Схожие клинические проявления имеет неонатальный прогероидный синдром (синдром Видемана-Раутенштрауха; в мире описано около 50 пациентов), который, в отличие от синдрома Хатчинсона-Гилфорда, манифестирует практически с рождения [1][3–4].
Несмотря на различный патогенез, для прогероидных синдромов характерно мультиорганное поражение: патология сердечно-сосудистой, нервной, эндокринной, пищеварительной, дыхательной, зубочелюстной систем, нарушения слуха и зрения, дерматологические проявления и др. Практически всегда имеет место поражение опорно-двигательного аппарата, проявляющееся деформациями позвоночника и конечностей, нарушением оссификации скелета, изменением структуры костной ткани, множественными контрактурами мелких и крупных суставов [1][5–7].
Под наблюдением находились 6 пациентов: 5 детей (3 мальчика и 2 девочки) с детской прогерией (синдром Хатчинсона-Гилфорда) и одна девочка с неонатальным прогероидным синдромом (синдром Видемана-Раутенштрауха).
Пациенты №1–4 имели классическую манифестацию синдрома Хатчинсона-Гилфодра в первые месяцы жизни, в 11 экзоне гена LMNA выявлен типичный для детской прогерии гетерозиготный патогенный вариант с.1824C>T. У пациентки №5 отмечена более ранняя манифестация заболевания с более тяжелыми фенотипическими проявлениями; выявленный в гене LMNA патогенный вариант c.1968+1G>A подтверждает неклассический тип синдрома Хатчинсона-Гилфорда (табл. 1).
Таблица 1. Характеристики пациентов с прогероидными синдромами
|
Пациент № |
Пол |
Заболевание |
Мутация |
Возраст манифестации |
Возраст постановки диагноза |
|
1 |
Девочка |
синдром Хатчинсона-Гилфорда |
c.1824C>T в гене LMNA |
2 мес. |
2 года 3 мес. |
|
2 |
Мальчик |
синдром Хатчинсона-Гилфорда |
c.1824C>T в гене LMNA |
9 мес. |
5 лет |
|
3 |
Мальчик |
синдром Хатчинсона-Гилфорда |
c.1824C>T в гене LMNA |
1 мес. |
1,5 года |
|
4 |
Мальчик |
синдром Хатчинсона-Гилфорда |
c.1824C>T в гене LMNA |
1 мес. |
1 год 4 мес. |
|
5 |
Девочка |
синдром Хатчинсона-Гилфорда |
c.1968+1G>A в гене LMNA |
с рождения |
1 год 6 мес. |
|
6 |
Девочка |
синдром Видемана-Раутенштрауха |
c.3337- 11T>C/ c.3677T>C в гене POLR3A |
с рождения |
6 лет 5 мес. |
У пациентки №5 прогероидные проявления отмечались с рождения, диагноз синдрома Видемана-Раутенштрауха был подтвержден в результате выявления в гене POLR3A компаунд-гетерозиготного патогенного варианта c.3337-11T>C/c.3677T>C [7].
Диагноз был установлен пациентам в возрасте 1,9 (1,5; 4,3) года (Ме (25%; 75%)). Возраст пациентов на момент первичного обследования — 6,0 (3,5; 7,2) лет, на момент повторного — 7,6 (7,5; 9,3) года.
Девочка, рожденная от физиологической беременности с нормальными росто-весовыми показателями (длина — 49 см (SDS -0,51), масса тела — 3200 г (SDS -0,58)) и отсутствием специфического фенотипа. Синдром Хатчинсона-Гилфорда был установлен в возрасте 2 лет 3 месяцев по результатам объективного осмотра и подтвержден молекулярно-генетическими методами исследования (табл. 1).
При первичном обследовании в возрасте 2-х лет 8 месяцев у девочки наблюдалась прогрессирующая задержка роста и дефицит массы тела, классический прогероидный фенотип (мраморные, сухие кожные покровы, выраженная венозная сеть на голове, выраженный периорбитальный цианоз, цианоз носогубного треугольника, генерализованная липодистрофия, клювовидный нос, микрогнатия, шарообразные глаза, экзофтальм, тотальная алопеция, отсутствие бровей), генерализованная липодистрофия, гепатопатия, дислипидемия. При осмотре ортопедом выявлены множественные контрактуры мелких суставов кистей, а также более крупных — тазобедренных, коленных, голеностопных суставов (табл. 2). По данным рентгенографии, отмечены изменения формы и структуры эпифизов и метафизов бедренных костей (рис. 1).
Таблица 2. Антропометрические показатели, ортопедические проявления и результаты рентгенологических исследований пациентов
|
Пациент |
Возраст обследования |
Рост, см (SDS роста) |
Вес, кг (SDS ИМТ) |
Ортопедические проявления |
Костный возраст (по Greulich-Pyle), лет |
Минеральная плотность костной ткани по данным денситометрии, Z-score |
|
1 |
2 года 8 мес. |
80 (-2,79) |
8,0 (-3,88) |
Контрактуры суставов (межфаланговых пальцев кистей, тазобедренных, коленных, голеностопных) |
- |
- |
|
5 лет 4 мес. |
91,5 (-3,66) |
10,0 (-3,25) |
Прогрессирование выявленных ранее контрактур; плоско-вальгусные стопы |
- |
- |
|
|
2 |
5 лет 6 мес. |
100 (-2,35) |
11,0 (-5,94) |
Контрактуры суставов (межфаланговых пальцев кистей, локтевых, тазобедренных, коленных); ограничение подвижности в лучезапястных суставах; плоско-вальгусные стопы |
4,5 |
- |
|
7 лет 5 мес. |
101 (-3,82) |
11,2 (-5,37) |
Прогрессирование выявленных ранее контрактур, формирование контрактур в лучезапястных суставах; плоско-вальгусные стопы |
- |
-3,1 |
|
|
3 |
7 лет 5 мес. |
100 (-4,05) |
12,1 (-3,50) |
Асептический некроз головки правой бедренной кости (болезни Легга-Кальве-Пертеса) 3 ст.; экструзия головки правой бедренной кости; coxa valga с 2х сторон; врожденный подвывих левого бедра; контрактуры суставов (локтевых, лучезапястных, пальцев кистей, тазобедренных, коленных, голеностопных, пальцев стоп) |
- |
- |
|
9 лет 3 мес. |
105,5 (-4,51) |
12,0 (-5,67) |
Без существенной динамики |
9,5 |
-2,9 |
|
|
4 |
9 лет 10 мес. |
Множественные контрактуры суставов; кифоз грудного отдела позвоночника, незаращение задних дужек LV, SI позвонков, деформация тел позвонков; диспластический тип строения крыш вертлужных впадин с латерализацией головок бедренных костей с тенденцией к подвывиху справа; «coxa valga» с двух сторон |
7 |
-3,8 |
||
|
11 лет 7 мес. |
Ранее выявленные изменения без существенной динамики; закрытый вывих левого бедра, кистовидная перестройка субхондральных отделов левой бедренной кости (гипсовая иммобилизация) |
- |
- |
|||
|
5 |
1 год 6 мес. |
68 (- 4,28) |
5,8 (- 4,73) |
Контрактуры суставов (пальцев кистей, лучезапястных, тазобедренных, коленных, голеностопных суставов); резкое ограничение движений в лучезапястных суставах, при нормальном объеме движений в локтевых суставах; гипоплазия ребер; кифоз грудного отдела позвоночника |
- |
- |
|
6 |
6 лет 5 мес. |
99 (-3,14) |
10,0 (-5,49) |
Контрактуры суставов (тазобедренных, коленных); диспластический тип строения позвоночника, нарушение осанки по сколиотическому типу |
- |
- |
|
7 лет 6 мес. |
103 (-3,41) |
10,4 (-6,20) |
Уменьшение контрактур в тазобедренных суставах, улучшение осанки |
6 |
- 3,8 |
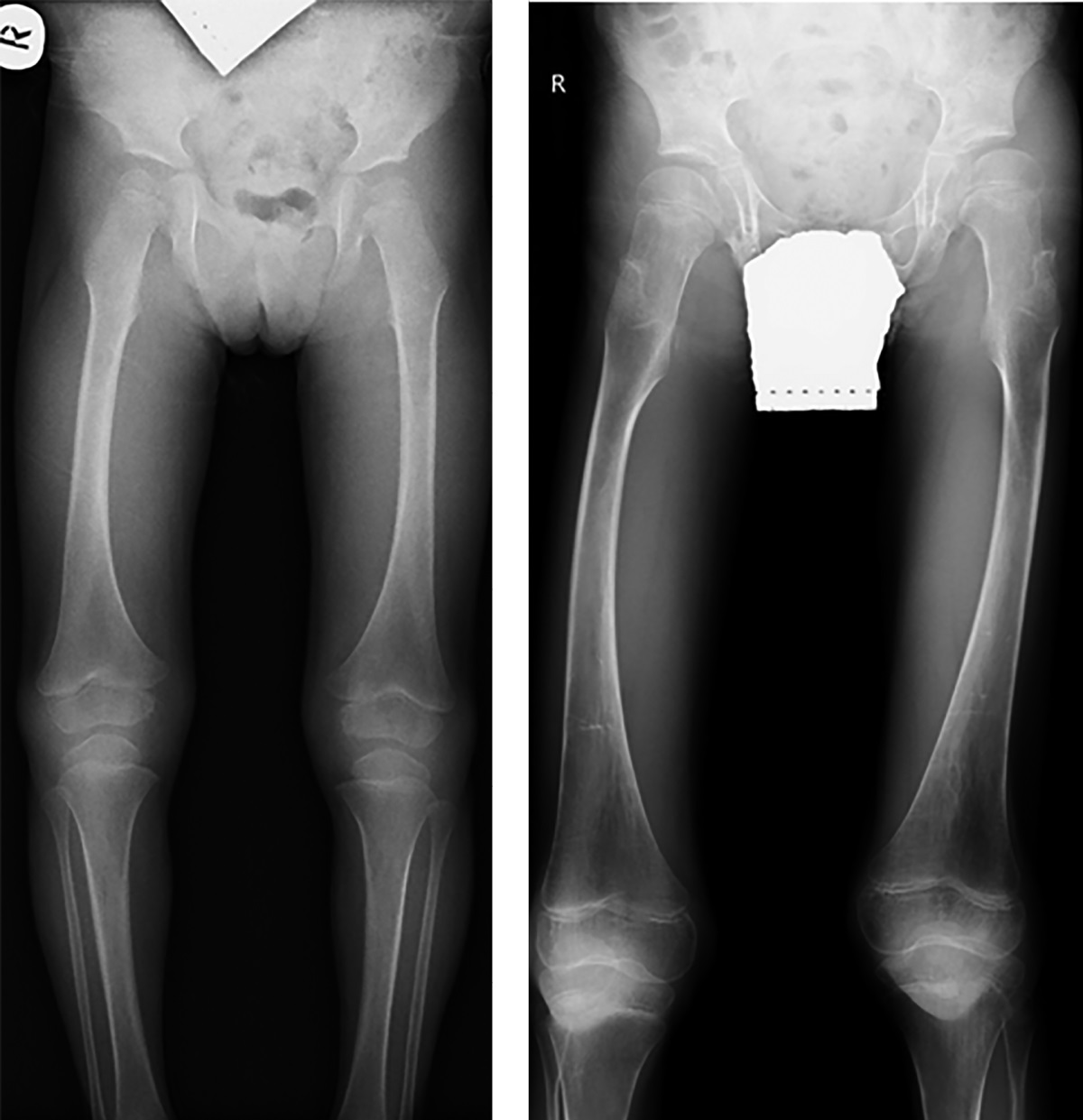
Рисунок 1. Пациентка №1, 2 года 8 мес., с синдромом Хатчинсона-Гилфорда.
Рентгенография таза с захватом нижних конечностей на протяжении. Форма и структура эпифизов и метафизов бедренных костей изменена.
Спустя 2,5 года, в возрасте 5 лет 4 месяцев, при осмотре фенотип девочки не изменился, однако задержка роста и дефицит массы тела стали более выражены. Отмечено прогрессирование ранее выявленных контрактур, а также формирование плоско-вальгусных стоп (табл. 2).
Мальчик, рожденный от физиологической беременности на 39‑й неделе с низко-нормальной массой тела (длина — 50 см (SDS -0,50), масса тела — 2800 г (SDS -1,85)) и отсутствием специфического фенотипа. После 9 месяца жизни наблюдалось выпадение волос и бровей, после 1 года жизни — изменение внешнего вида. Синдром Хатчинсона-Гилфорда был установлен в возрасте 5 лет и подтвержден молекулярно-генетическими методами исследования (табл. 1).
Первичное обследование было проведено в возрасте 5 лет 6 месяцев. Наблюдалась тяжелая задержка роста и дефицит массы тела, классические прогероидные черты лица (тонкая мраморная сухая гиперпигментированная кожа, выраженная венозная сеть на голове, нижних конечностях, выраженный периорбитальный цианоз, цианоз носогубного треугольника, треугольное лицо, клювовидный нос, микрогнатия, шарообразные глаза, экзофтальм, пушковый волос на голове, редкие брови, ресницы), генерализованная липодистрофия. При осмотре ортопеда отмечались выраженные контрактуры суставов кистей, в том числе пальцев; ограничение подвижности в лучезапястных суставах; разгибательные контрактуры в локтевых суставах; сгибательные контрактуры в коленных суставах; плоско-вальгусные стопы. По результатам обследования отмечалось незначительное отставание костного возраста, изменения формы и структуры эпифизов и метафизов бедренных костей (табл. 2, рис. 2).
Спустя 2 года, в 7 лет 5 месяцев, при осмотре наблюдалось прогрессирующее снижение росто-весовых показателей и деформации тазобедренных суставов, при этом фенотип пациента не изменился. Рентгенологически отмечено нарастание снижения плотности костной ткани метаэпифизарных отделов, преимущественно бедренных костей. С целью оценки минеральной плотности костной ткани была проведена денситометрия, по результатам которой был диагностирован остеопороз (табл. 2).
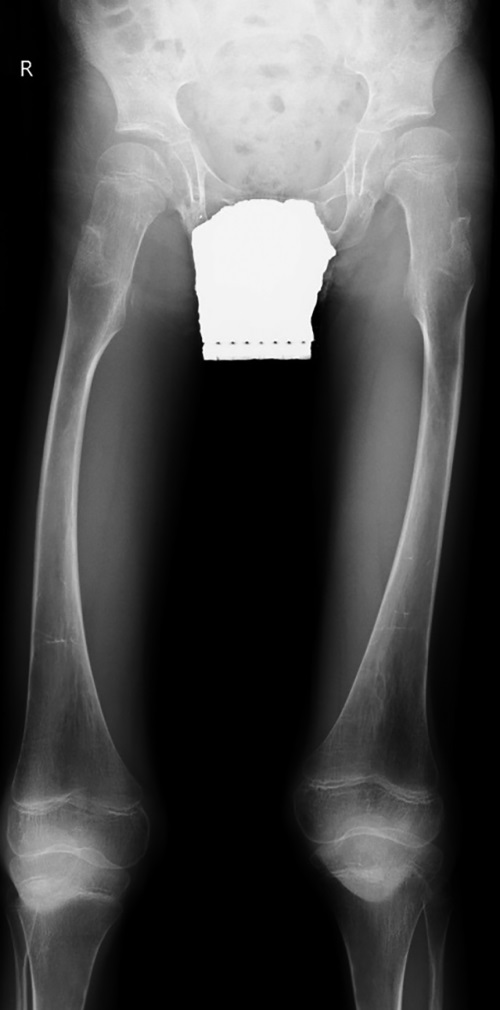
Рисунок 2. Пациент №2, 5 лет 6 мес., с синдромом Хатчинсона-Гилфорда.
Рентгенография таза с захватом нижних конечностей на протяжении. Выраженные изменения формы и структуры эпифизов и метафизов бедренных костей.
Мальчик, рожденный от первой беременности на 37‑й неделе гестации с нормальными росто-весовыми показателями (масса тела — 3030 г (SDS -1,27), длина — 50 см (SDS -0,38)) и отсутствием клинических проявлений заболевания. После 1 месяца жизни у мальчика стали отмечаться уплотнения под кожей голеней, гиперпигментация кожи на животе; после 1 года жизни — выпадение волос, резкая задержка массово-ростовых показателей, изменение внешнего вида (рис. 3); повышение печеночных ферментов (АЛТ, АСТ) в 3 раза. В 1,5 года, по результатам молекулярно-генетического исследования, мальчику был установлен диагноз: «Классический синдром Хатчинсона-Гилфорда» (табл. 1).
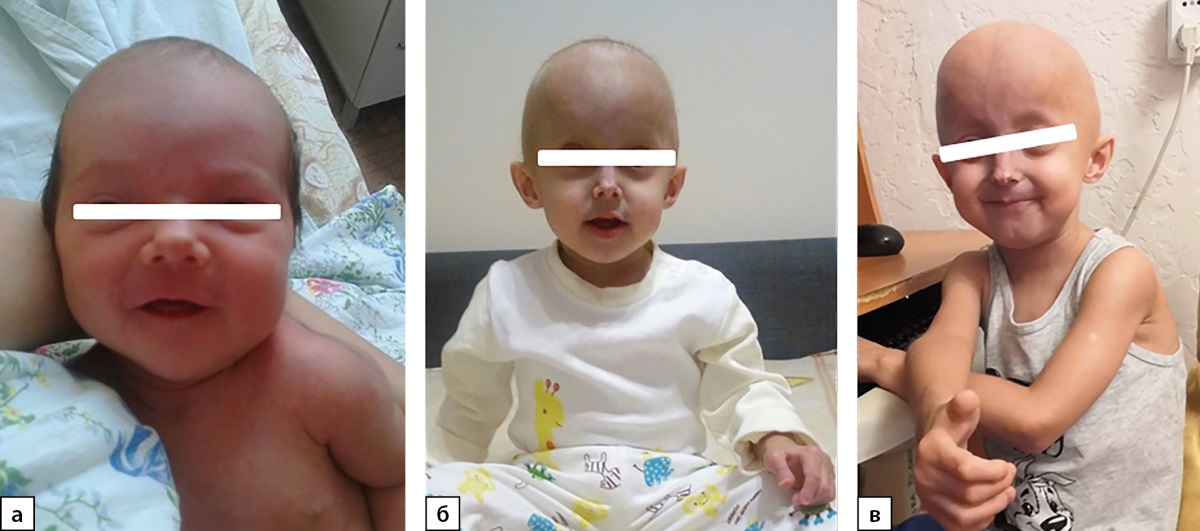
Рисунок 3. Пациент №3: а) на 4‑е сутки жизни; б) в 1,5 года; в) в 4 года.
Впервые пациент был осмотрен в возрасте 7 лет 5 месяцев, наблюдалась тяжелая задержка роста, дефицит массы тела, генерализованная липодистрофия, фенотипические изменения (мраморная сухая кожа, участки гипер- и гипопигментации, выражена венозная сеть на голове, изменение формы лицевого и мозгового черепа, клювовидный нос, микрогнатия, шарообразные глаза, экзофтальм, тотальная алопеция, отсутствие бровей).
При объективном осмотре ортопеда и рентгенологическом обследовании выявлены асептический некроз головки правой бедренной кости (болезни Легга-Кальве-Пертеса) 3 ст; экструзия головки правой бедренной кости; coxa valga с двух сторон; врожденный подвывих левого бедра (рис. 4). Также имели место множественные контрактуры локтевых, лучезапястных; тазобедренных, коленных, голеностопных суставов, сочетающиеся с деформациями контрактурами суставов пальцев кистей и стоп (табл. 2).
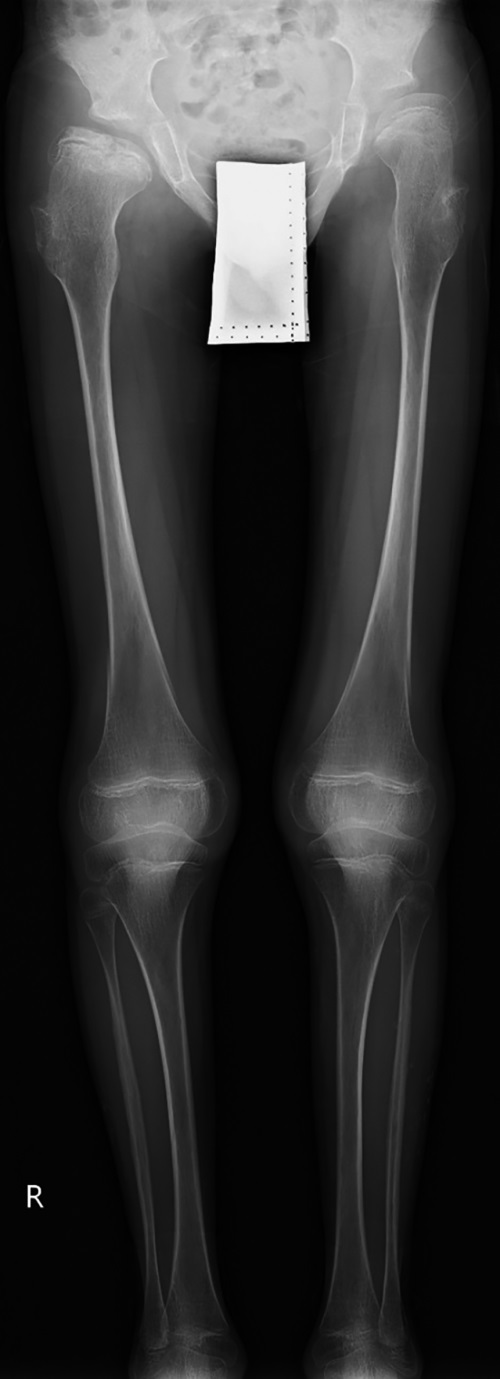
Рисунок 4. Пациент №3, 7 лет 5 мес., с синдромом Хатчинсона-Гилфорда.
Рентгенография таза с захватом нижних конечностей на протяжении. Выраженные изменения формы и структуры эпифизов и метафизов бедренных костей; болезнь Легга-Кальве-Пертеса 3 ст.; экструзия головки правой бедренной кости; coxa valga с двух сторон; врожденный подвывих левого бедра.
При осмотре в возрасте 9 лет 3 месяцев отмечено прогрессирующее снижение росто-весовых показателей, сохранение специфического фенотипа, ортопедические проявления без существенной динамики. При рентгенографии кистей рук выявлено незначительное опережение костного возраста, по результатам денситометрии — остеопороз (табл. 2).
Мальчик, рожденный от первой беременности, протекавшей на фоне ОРВИ в 1 и 2 триместрах, токсикоза до 2 триместра, умеренных отеков в 3 триместре, на 38‑й неделе гестации с нормальными росто-весовыми показателями (длина — 51 см (SDS: +0,82), масса — 2970 (SDS: -0,55) и отсутствием специфического фенотипа. При рождении состояние тяжелое за счет дыхательной недостаточности 3 ст. на фоне перенесенной перинатальной асфиксии и аспирации околоплодных вод, двое суток находился на искусственной вентиляции легких (ИВЛ).
С первого месяца жизни было отмечено снижение темпов роста и набора веса, с 3–4 месяца — проявления специфического фенотипа. В 1 год 4 месяца, по результатам молекулярно-генетического исследования, мальчику был установлен диагноз: «Классический синдром Хатчинсона-Гилфорда».
Первичное обследование было проведено в возрасте 9 лет 10 месяцев, при объективном осмотре отмечалась тяжелая задержка роста и дефицит массы тела, генерализованная липодистрофия, характерные фенотипические изменения. При осмотре ортопедом выявлены множественные контрактуры суставов; кифоз грудного отдела позвоночника, незаращение задних дужек LV, SI позвонков, деформация тел позвонков; диспластический тип строения крыш вертлужных впадин с латерализацией головок бедренных костей с тенденцией к подвывиху справа; «coxa valga» с двух сторон, что также наблюдалось у пациента №3.
В возрасте 11 лет 7 месяцев при падении во время игры на детской площадке диагностирован закрытый вывих левого бедра, кистовидная перестройка субхондральных отделов левой бедренной кости, проведена гипсовая иммобилизация; ранее выявленные изменения без существенной динамики.
Девочка, рожденная от 5‑й беременности на 38‑й неделе гестации с дефицитом массы тела (длина — 49 см (SDS -0,92), масса тела — 2505 г (SDS -2,62)). На 2–3 месяце жизни отмечено снижение темпов роста и набора веса, формирование специфического фенотипа (мраморность кожных покровов, шелушение заушной области, кольцевидная эритема (1,7х1,7 см) на волосистой части головы и спины, плотные отеки в ягодичной области, «птичье лицо», гипоплазия орбит, проптоз глазных яблок, узкий клювовидный нос, микростомия, микрогнатия, готическое небо, диспластичные ушные раковины.
По данным рентгенографии и МСКТ, визуализированы расширенные большой и малые роднички и аномалии строения затылочной кости (рис. 5).
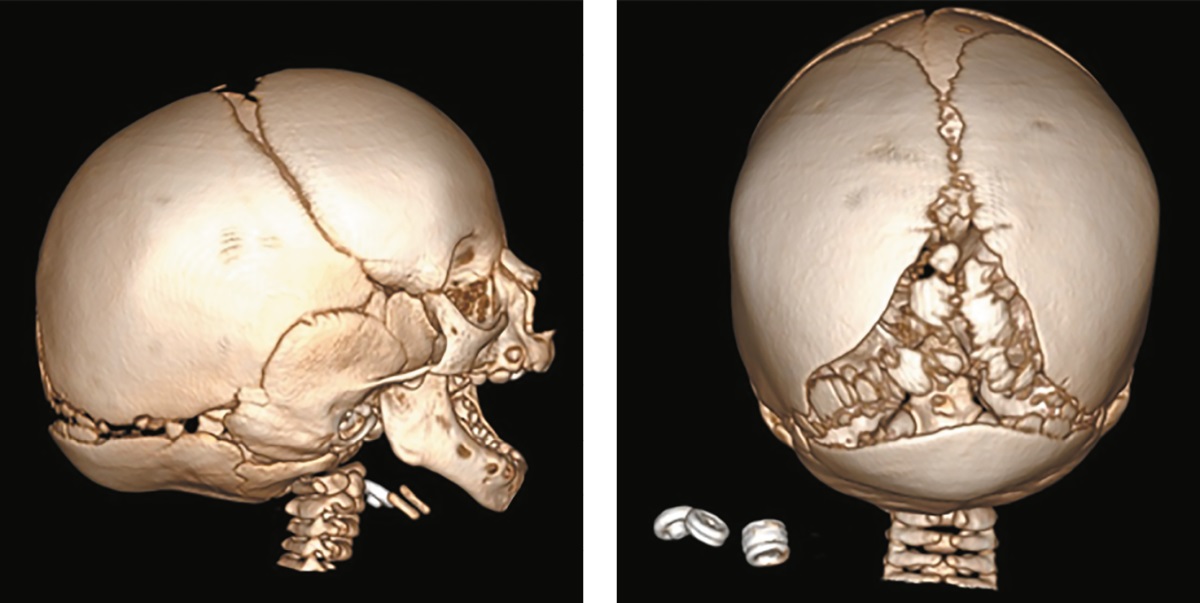
Рисунок 5. Пациентка №4., 1 год 6 мес. с тяжелой формой синдрома Хатчинсона-Гилфорда.
МСКТ черепа 3D-реконструкция. Мультифрагментарная фрагментация вдоль краниальных швов.
При этом имели место маленькие кисти и стопы, а также начальные проявления липодистрофии.
В возрасте 1 года 6 месяцев девочке был подтвержден неклассический тип синдрома Хатчинсона-Гилфорда (табл. 1). При объективном осмотре отмечалась задержка роста, дефицит массы тела, проявления специфического фенотипа (тонкие мраморные кожные покровы с единичными участками гиперпигментации и выраженным венозным рисунком, клювовидный нос, микрогнатия, шарообразные глаза, тотальная аллопеция, диспластичные ушные раковины, маленькие кисти и стопы, участки липодистрофии).
При осмотре ортопедом наблюдались сгибательно-разгибательные контрактуры пальцев кистей и лучезапястных суставов, резкое ограничение движений в лучезапястных суставах при нормальном объеме движений в локтевых суставах (табл. 2). Наряду с этим имелись сгибательные контрактуры в тазобедренных суставов и выраженные, около 150 градусов, сгибательные контрактуры в коленных суставах. На рентгенограмме таза и тазобедренных суставов нарушения соотношения и признаков асептического некроза выявлено не было (рис. 6).
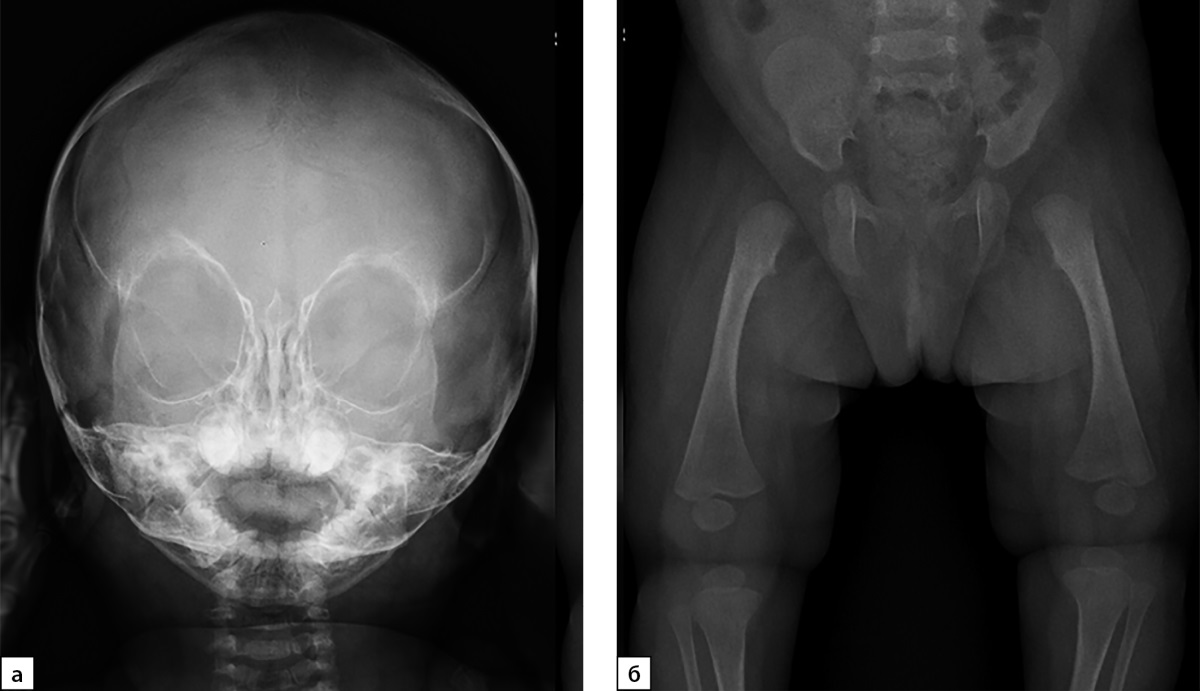
Рисунок 6. Пациентка №4, а — рентгенограмма черепа в прямой проекции. Вид лицевого скелета; б — тазобедренные суставы: гипоплазия костей таза. Проксимальные эпифизы бедренных костей не оссифицированы.
Аналогичные изменения в виде сгибательных контрактур имели место в голеностопных суставах. Со стороны костного скелета грудной клетки и позвоночника обращали на себя внимание гипоплазия ребер выраженное изменение статики позвоночника в виде кифоза грудного отдела (табл. 2).
Девочка с неонатальным прогероидным синдромом Видемана-Раутенштрауха обследована ортопедом-травматологом в 6 лет 4 месяца и 7 лет 6 месяцв (табл. 2).
Известно, что при рождении на сроке 37 недель гестации выявлена задержка внутриутробного развития (длина — 46 см (SDS -2,20), масса тела — 1840 г (SDS -4,27)), которая наблюдалась с 24‑й недели гестации. Также отмечено наличие псевдогидроцефалии, прогероидных черт лица, генерализованной липодистрофии, неонатального резца верхней челюсти, выраженной мышечной гипотонии. О каких-либо костно-суставных нарушениях на момент рождения достоверных данных в медицинской документации нет.
В 6 лет 5 месяцев, при первичном обследовании выявлено прогрессирование выраженности (по сравнению с показателями при рождении) задержки роста, тяжелый дефицит массы тела (табл. 2), классические фенотипические прогероидные черты (псевдогидроцефальная форма черепа, выступающие массивные лобные бугры, треугольное лицо с гипоплазией скуловой кости, клювовидный нос, микрогнатия, адентия; периорбитальный цианоз, выраженная венозная сеть на голове, нижних конечностях; генерализованная липодистрофия с изолированными участками перераспределения подкожно-жировой клетчатки; гипотрихоз, редкие брови и ресницы, эктопион верхнего века, длинный язык). Из анамнеза известно, что отмечалось позднее закрытие большого родничка (после 4,5 года). (рис. 7).
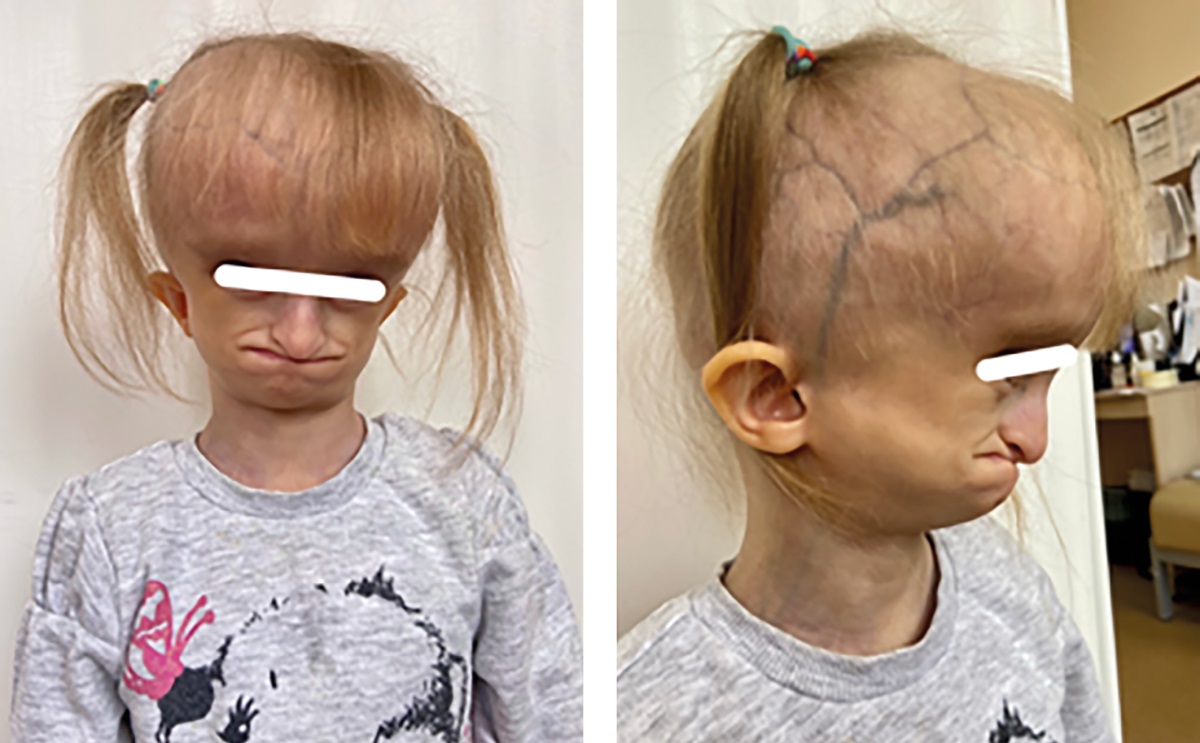
Рисунок 7. Пациентка №5 в возрасте 6 лет 5 месяцев [8].
При объективном осмотре ортопедом выявлен диспластический тип строения позвоночника, нарушение осанки по сколиотическому типу (угроза формирования S-образного сколиоза), сгибательные контрактуры тазобедренных и коленных суставов.
При повторном осмотре через год, в возрасте 7,5 года, отмечалось нарастание задержки роста и прогрессия дефицита массы тела; внешний вид пациентки существенно не изменился. По результатам проведенного обследования выявлено незначительное отставание костного возраста (костный возраст — 6 лет), остеопороз (Z-score: -3,8). При объективном осмотре ортопеда отмечалось уменьшение контрактур в тазобедренных суставах, улучшение осанки (табл. 2).
Синдром Хатчинсона-Гилфорда (детская прогерия) — это ультраредкое наследственное заболевание, характеризующееся ускоренным старением организма и высокой частотой летальности в детском возрасте. Заболевание имеет аутосомно-доминантный тип наследования и в 90% случаев вызван гетерозиготной мутацией c.1824C>T в кодоне 608, в пределах экзона 11 гена LMNA (расположен на хромосоме 1q22), определяющей классический фенотип заболевания. В 10% случаев описана неклассическая форма прогерии, которая обусловлена другими патогенными вариантами в гене LMNA: c.1822G>A, c.1821G>A, c.1968G>A, c.1968+1G>C, c.1968+1G>A, c.1968+2T>A, c.1968+2T>C, c.1968+5G>A, c.1968+5G>C. Неклассическая форма прогерии приводит к чрезмерному накоплению прогерина в клетке и проявляется клиникой синдрома Хатчинсона-Гилфорда с менее или более выраженными проявлениями по сравнению с типичным фенотипом [1][3].
При рождении у всех пациентов отмечаются нормальные росто-весовые показатели, отсутствует специфический фенотип. Клинически заболевание проявляется в первые 18 месяцев жизни и характеризуется снижением темпов роста и прибавки массы тела, прогрессирующей алопецией, «склеродермоподобными» изменениями кожи, потерей подкожно-жировой клетчатки, отсроченным сращением черепных швов, проявлением характерного внешнего вида (преобладание мозгового отдела черепа над лицевым; тонкая морщинистая гиперпигментированная кожа с выступающим венозным рисунком; микрогнатия верхней и нижней челюсти, синюшность носогубного треугольника, узкий клювовидный нос, тонкая верхняя и нижняя губа, маленький рот, позднее прорезывание и скученность зубов) [2][9–11].
С течением жизни у пациентов с прогерией присоединяется патология сердечно-сосудистой, зубочелюстной систем, дерматологические проявления, возможны нарушения зрения и слуха. Одним из ключевых клинических проявлений является патология опорно-двигательного аппарата и костной ткани [1].
Костно-суставные изменения при синдроме Хатчинсона-Гилфорда, как правило, включают в себя формирование контрактур больших и малых суставов с прогрессирующей ригидностью, что впоследствии ведет к нарушению объема движений в суставе. Так, Melissa A. Merideth, Leslie B. Gordon и др. в 2008 г. выявили у 100% пациентов с детской прогерией нарушение диапазона движения в трех периферических суставах: средний диапазон движений при вращении запястья, лодыжки и бедра составлял 63 градуса (нормальное значение — 150), 36 градусов (нормальное значение — 70) и 69 градусов (нормальное значение — 90) соответственно; сгибание позвоночника было снижено у 73% обследованных детей [12].
Известно, что для пациентов с прогерией характерно развитие вывиха бедра и возникновение асептических некрозов бедренных костей по типу болезни Легга-Кальве-Пертеса, что, по данным обзора литературы, обусловлено нарушениями структуры костной ткани, деформацией метафизов и эпифизов и прогрессирующей coxa valga проксимального отдела бедренных костей [1].
Костный возраст при синдроме Хатчинсона-Гилфорда, как правило, соответствует хронологическому и имеет незначительную тенденцию к опережению [1][13].
При проведении стандартной денситометрии поясничного отдела позвоночника (с отсутствием коррекции результатов денситометрии на хронологические возраст, рост и костный возраст пациента) у 100% пациентов отмечается снижение минеральной плотности костной ткани (показатель Z-score ≤-2,0 стандартных отклонения (SD)). В 2011 г. Catherine M. Gordon, Leslie B. Gordon и др. отмечали, что при изменении результата с учетом роста и возраста Z- score повышается как для позвоночника, так и для всего тела на + 1,24 и + 1,71 SD соответственно, а риск переломов не превышает рисков здоровых детей. В настоящее время вопрос остается дискутабельным [14].
Биохимические показатели костного метаболизма, такие как остеокальцин, N-телопептид, кальций, фосфор, щелочная фосфатаза, общий белок, паратиреоидный гормон, 25(ОН)D не отличаются от такового для здоровых детей соответствующего возраста [14].
Неонатальный прогероидный синдром (синдром Видемана-Раутенштрауха или неонатальный прогероидный синдром) — ультраредкое наследственное заболевание, характеризующееся врожденной генерализованной липодистрофией и прогероидными чертами лица. Заболевание имеет аутосомно-рецессивный тип наследования и связано с биаллельными вариантами в генах POLR3A, POLR3B и POLR3GL [4][15].
Клинически заболевание проявляется еще внутриутробно, характеризуясь задержкой темпов роста, выраженным отставанием бипариетального (расстояние от одной височной области до другой) и брюшного размеров после 18–20 недели гестации. При рождении у детей отмечается специфический фенотип (псевдогидроцефалия с широким передним родничком, тонкой кожей, ярко выраженными венами скальпа и тонкими редкими волосами; треугольное лицо с тонкой синюшной морщинистой и гиперпигментированной кожей; клювовидный нос, низко посаженные уши, микрогнатия, 2–4 неонатальных резца; генерализованная липодистрофия с единичными участками подкожно-жировой клетчатки (ПЖК), локализующуюся в щечной, грудной, ягодичной, поясничной областях), прогрессирующая задержка роста и темпы набора массы тела [5–7].
С течением жизни сохраняется прогрессирующая задержка роста и тяжелый дефицит веса. После 1,5–2 лет у пациентов отмечаются патологические изменения опорно-двигательного аппарата: формирование контрактур больших и малых суставов, прогрессирующий кифосколиоз. Костный возраст, как правило, имеет тенденцию к отставанию от паспортного более, чем на 2 года [16].
При проведении денситометрии поясничного отдела позвоночника у пациентов с неонатальным прогероидным синдромом, как и у пациентов с синдромом Хатчинсона-Гилфорда, отмечается снижение минеральной плотности костной ткани (показатель Z-score ≤-2,0 стандартных отклонения (SD), однако высокой частоты переломов у пациентов не отмечается [16].
Изменения опорно-двигательного аппарата у всех наблюдаемых нами пациентов были представлены типичными для прогероидных синдромов контрактурами межфаланговых пальцев кистей и стоп, лучезапястных, локтевых, тазобедренных, коленных и голеностопных суставов и изменениями позвоночника в виде кифоза и нарушения осанки по сколиотическому типу, выявленными у половины пациентов (№4, 5, 6). Данные изменения были диагностированы при первом обследовании, которое проводилось через 0,5 (0,1; 4,6) лет после молекулярно-генетического подтверждения диагноза.
При повторном обследовании только у пациентки с неонатальным прогероидным синдромом (№6) отмечалась положительная динамика в виде уменьшения контрактур в тазобедренных суставах и улучшения осанки. У пациентов с детской прогерией младшего возраста (№1 и 2) отмечено прогрессирование контрактур в период наблюдения.
У двух пациентов более старшего возраста (№3 и 4) за период наблюдения контрактуры суставов без существенной динамики; по тяжести поражения на первое место выходят характерные для детской прогерии изменения тазобедренных суставов формирование coxa valga с развитием асептического некроза головки бедренной кости (пациент №3) и закрытого вывиха левой бедренной кости (пациент №4), что требует особого внимания врача-ортопеда с целью своевременной коррекции.
В качестве особенностей пациентов, у которых заболевание манифестировало с рождения (пациент №5 и №6), обращает на себя внимание раннее развитие сгибательных контрактур больших суставов (коленных, тазобедренных, голеностопных), раннее формирование кифоза и сколиоза, позднее закрытие большого родничка (пациент №6).
Полученные нами результаты определения костного возраста согласуются с данными литературы: костный возраст либо соответствовал хронологическому (пациент №3), либо отставал на 18 (15; 26) месяцев.
При проведении денситометрии 4 пациентам (№2, 3, 4 ,6) в возрасте 8,3 (7,5; 9,1) года у всех выявлен остеопороз — Z-критерий: -3,4 (-3,0; -3,8). Однако переломы в нашей группе выявлены не были, что также согласуется с литературными данными.
Российский клинический опыт ведения пациентов с прогероидными синдромами на сегодняшний день остается фрагментарным и представлен единичными публикациями. Так, продемонстрированная в данной статье девочка является первой описанной в РФ пациенткой с синдромом Видемана-Раутенштрауха, что подчеркивает уникальность данного заболевания [8].
Описание пациентов с синдромом Хатчинсона-Гилфорда в России ограничивается 2 публикациями. В 1995 г. Фофанова О.В. описала девочку 3 лет 9 месяцев с верифицированным диагнозом синдрома Хатчинсона-Гилфорда, где также сообщала о выраженных патологических изменениях опорно-двигательного аппарата в виде двустороннего врожденного вывиха и дисплазии тазобедренных суставов, утолщения межфаланговых суставов и концевых фаланг кистей и стоп; варусной девиации верхней трети предплечий [17].
Спустя почти три десятилетия, в 2022 г., коллектив авторов из Санкт-Петербурга и Казахстана описал девочку с детской прогерией в возрасте 9 лет 3 месяцев. При объективном осмотре костно-суставной системы отмечалось ограничение движений в коленных и тазобедренных суставах за счет фиброзно-склеротических изменений; контрактуры коленных, голеностопных и межфаланговых суставов кистей рук; воронкообразная деформация грудной клетки. [18].
На сегодняшний день известно, что к прогероидным синдромам относятся не только синдром Видемана-Раутенштрауха и синдром Хатчинсона-Гилфорда, но и рестриктивная дермопатия, мандибулоакральная дисплазия с липодистрофией типов А и В, синдромы Вернера, Блума, Ротмунда-Томсона, пигментная ксеродерма, синдром Коккейна, трихотиодистрофия и другие прогероидные синдромы с признаками преждевременного старения — врожденный дискератоз, синдром Пенттинена и т.п. Для данных синдромов также характерны костно-суставные изменения [1–2].
В 2021 г. Belaya Z. и соавт. описали пациента 24 лет с подтвержденным молекулярно-генетическими методами исследования диагнозом «Врожденный дискератоз» и рецедивирующими двусторонними малотравматичными переломами бедра, беспокоившие его в течение 10 лет. Для предотвращения дальнейших переломов была инициирована терапия золедроновой кислотой в дозе 5 мг, продемонстрировавшая клиническую эффективность в течение четырех лет наблюдений. На сегодняшний день это единственный опыт применения бисфосфонатов у пациента с синдромом преждевременного старения в РФ и может рассматриваться в качестве возможной терапии остеопороза у пациентов с прогероидными синдромами [19].
Таким образом, все выявленные у наблюдаемых нами пациентов изменения костной ткани и опорно-двигательного аппарата соответствуют особенностям, описанным при синдромах Хатчинсона-Гилфорда и Видемана-Раутенштрауха.
По данным наших наблюдений, при неклассической форме детской прогерии отмечаются более ранние изменения опорно-двигательного аппарата по сравнению с классической формой, что подтверждает тяжелое течение заболевания при данном генотипе. Напротив, изменения у пациентки с неонатальным прогероидным синдромом более схожи с изменениями при классической детской прогерии, что свидетельствует о схожести фенотипов синдромов преждевременного старения.
Источники финансирования. Работа выполнена по инициативе авторов без привлечения финансирования.
Конфликт интересов. Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с содержанием настоящей статьи.
Согласие пациентов. Пациенты добровольно подписали согласие на публикацию фотографий и персональной медицинской информации в обезличенной форме в журнале «Остеопороз и остеопатии».
1. Gordon LB, Brown WT, Collins FS. Hutchinson-Gilford Progeria Syndrome. 2003 Dec 12 [updated 2023 Oct 19]. In: Adam MP, Feldman J, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993–2024
2. Голоунина О.О., Фадеев В.В., Белая Ж.Е. Наследственные синдромы с признаками преждевременного старения. // Остеопороз и остеопатии. — 2019. — Т.22. — №3. — С. 4-18. https://doi.org/10.14341/osteo12331
3. Ferreira-Marques M, Carvalho A, Franco AC, Leal A, Botelho M, et al. Ghrelin delays premature aging in Hutchinson-Gilford progeria syndrome. Aging Cell. 2023;22(12):e13983. https://doi.org/10.1111/acel.13983
4. Wambach JA, Wegner DJ, Patni N, Kircher M, Willing MC, et al. Bi-allelic POLR3A Loss-of-Function Variants Cause Autosomal-Recessive Wiedemann-Rautenstrauch Syndrome. Am J Hum Genet. 2018;103(6):968-975. https://doi.org/10.1016/j.ajhg.2018.10.010
5. Bitoun P, Lachassine E, Sellier N, Sauvion S, Gaudelus J. The Wiedemann-Rautenstrauch neonatal progeroid syndrome: a case report and review of the literature. Clin Dysmorphol. 1995;4(3):239-45
6. Rabah M. Shawky, Heba Salah Abd-Elkhalek, Shimaa Gad, Neveen S. Seifeldin. Neonatal progeroid syndrome (Wiedemann-Rautenstrauch syndrome) in an Egyptian child with premature loss of teeth, and café au lait skin patches. Egyptian Journal of Medical Human Genetics. 2012;13(2):227-231
7. Toriello HV. Wiedemann-Rautenstrauch syndrome. J Med Genet. 1990;27(4):256-7. https://doi.org/10.1136/jmg.27.4.256
8. Кунгурцева А.Л., Попович А.В., Тихонович Ю.В., Витебская А.В. Синдром Видемана-Раутенштрауха. Первое описание клинического случая в Российской Федерации. // Проблемы Эндокринологии. — 2024. — Т.70. — №2. — С. 86-93. https://doi.org/10.14341/probl13369
9. Cisneros B, García-Aguirre I, De Ita M, Arrieta-Cruz I, Rosas-Vargas H. Hutchinson-Gilford Progeria Syndrome: Cellular Mechanisms and Therapeutic Perspectives. Arch Med Res. 2023;54(5):102837. https://doi.org/10.1016/j.arcmed.2023.06.002
10. Batista NJ, Desai SG, Perez AM, Finkelstein A, Radigan R, et al. The Molecular and Cellular Basis of Hutchinson-Gilford Progeria Syndrome and Potential Treatments. Genes (Basel). 2023;14(3):602. https://doi.org/10.3390/genes14030602
11. Gordon LB, Harten IA, Patti ME, Lichtenstein AH. Reduced adiponectin and HDL cholesterol without elevated C-reactive protein: clues to the biology of premature atherosclerosis in Hutchinson-Gilford Progeria Syndrome. J Pediatr. 2005;146(3):336-41. https://doi.org/10.1016/j.jpeds.2004.10.064
12. Merideth MA, Gordon LB, Clauss S, Sachdev V, Smith AC, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med. 2008;358(6):592-604. https://doi.org/10.1056/NEJMoa0706898
13. Tsai A, Johnston PR, Gordon LB, Walters M, Kleinman M, Laor T. Skeletal maturation and long-bone growth patterns of patients with progeria: a retrospective study. Lancet Child Adolesc Health. 2020;4(4):281-289. https://doi.org/10.1016/S2352-4642(20)30023-7
14. Gordon CM, Gordon LB, Snyder BD, Nazarian A, Quinn N, et al. Hutchinson-Gilford progeria is a skeletal dysplasia. J Bone Miner Res. 2011;26(7):1670-9. https://doi.org/10.1002/jbmr.392
15. Temel SG, Ergoren MC, Manara E, Paolacci S, Tuncel G, Gul S, Bertelli M. Unique combination and in silico modeling of biallelic POLR3A variants as a cause of Wiedemann-Rautenstrauch syndrome. Eur J Hum Genet. 2020;28(12):1675-1680. https://doi.org/10.1038/s41431-020-0673-1
16. Dinleyici EC, Tekin N, Dinleyici M, Aksit MA. Clinical and laboratory findings of two newborns with Wiedemann-Rautenstrauch syndrome: additional features, evaluation of bone turnover and review of the literature. J Pediatr Endocrinol Metab. 2008;21(6):591-6
17. Фофанова О.В. Синдром Гетчинсона-Гилфорда (прогерия). // Проблемы Эндокринологии. — 1995. — Т.41. — №4. — С. 24-26. i: https://doi.org/10.14341/probl11459
18. Бучинская Н.В., Ахенбекова А.Ж., Бугыбай А.А., Костик М.М. Прогерия (синдром Хатчинсона – Гилфорда): обзор литературы и клинический случай. // Вопросы современной педиатрии. — 2022. — Т. 21. — №3. — С.253-264. oi: https://doi.org/10.15690/vsp.v21i3.2431
19. Belaya Z, Golounina O, Nikitin A, Tarbaeva N, Pigarova E, et al. Multiple bilateral hip fractures in a patient with dyskeratosis congenita caused by a novel mutation in the PARN gene. Osteoporos Int. 2021;32(6):1227-1231. https://doi.org/10.1007/s00198-020-05758-6
Кунгурцева Анастасия Леонидовна
119991, г. Москва, ул. Трубецкая, д. 8, стр. 2
Тихонович Юлия Викторовна, к.м.н.
г. Москва
Кульгускин Илья Юрьевич
г. Москва
Наталия Александровна Шолохова, д.м.н.
г. Москва
Максимова Юлия Владимировна, д.м.н.
г. Новосибирск
Витебская Алиса Витальевна, к.м.н.
г. Москва
|
|
1. Рисунок 1. Пациентка №1, 2 года 8 мес., с синдромом Хатчинсона-Гилфорда. Рентгенография таза с захватом нижних конечностей на протяжении. Форма и структура эпифизов и метафизов бедренных костей изменена. | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(626KB)
|
Метаданные ▾ | |
|
|
2. Рисунок 2. Пациент №2, 5 лет 6 мес., с синдромом Хатчинсона-Гилфорда. Рентгенография таза с захватом нижних конечностей на протяжении. Выраженные изменения формы и структуры эпифизов и метафизов бедренных костей. | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(305KB)
|
Метаданные ▾ | |
|
|
3. Рисунок 3. Пациент №3: а) на 4-е сутки жизни; б) в 1,5 года; в) в 4 года. | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(649KB)
|
Метаданные ▾ | |
|
|
4. Рисунок 4. Пациент №3, 7 лет 5 мес., с синдромом Хатчинсона-Гилфорда. Рентгенография таза с захватом нижних конечностей на протяжении. Выраженные изменения формы и структуры эпифизов и метафизов бедренных костей; болезнь Легга-Кальве-Пертеса 3 ст.; экструзия головки правой бедренной кости; coxa valga с двух сторон; врожденный подвывих левого бедра. | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(403KB)
|
Метаданные ▾ | |
|
|
5. Рисунок 5. Пациентка №4., 1 год 6 мес. с тяжелой формой синдрома Хатчинсона-Гилфорда. МСКТ черепа 3D-реконструкция. Мультифрагментарная фрагментация вдоль краниальных швов. | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Посмотреть
(558KB)
|
Метаданные ▾ | |
|
|
6. Рисунок 6. Пациентка №4, а — рентгенограмма черепа в прямой проекции. Вид лицевого скелета; б — тазобедренные суставы: гипоплазия костей таза. Проксимальные эпифизы | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(544KB)
|
Метаданные ▾ | |
|
|
7. Рисунок 7. Пациентка №5 в возрасте 6 лет 5 месяцев [8]. | |
| Тема | ||
| Тип | Материалы исследования | |
Посмотреть
(837KB)
|
Метаданные ▾ | |
Кунгурцева А.Л., Тихонович Ю.В., Кульгускин И.Ю., Шолохова Н.А., Максимова Ю.В., Витебская А.В. Особенности опорно-двигательного аппарата и костной ткани у пациентов с синдромами преждевременного старения. Остеопороз и остеопатии. 2025;28(3):34-45. https://doi.org/10.14341/osteo13204
Kungurtseva A.L., Tikhonovich Y.V., Kulguskin I.Y., Sholokhova N.A., Maksimova J.V., Vitebskaya A.V. Features of musculoskeletal apparatus and bone tissue in patients with premature aging syndromes. Osteoporosis and Bone Diseases. 2025;28(3):34-45. (In Russ.) https://doi.org/10.14341/osteo13204
117036, Москва, ул. Дмитрия Ульянова, д. 11.
Обработка персональных данных































