



ОРИГИНАЛЬНЫЕ ИССЛЕДОВАНИЯ
ОБОСНОВАНИЕ. Территории Арктической зоны Российской Федерации имеют риск развития дефицита 25-OH витамина D, который усугубляется низким уровнем инсоляции и обедненностью продуктов питания витаминами и минералами. Наряду с низким уровнем инсоляции и снижением концентрации витамина D в периоды с минимальной продолжительностью светового дня возможны проявления сезонного аффективного расстройства, характеризующегося нарушением циркадных ритмов и моноаминергических функций нейромедиаторов (серотонина, дофамина, норадреналина). Научная проблема заключается в оценке обеспеченности населения 25-OH витамином D и биогенными аминами и обнаружении ассоциативных изменений уровней 25-OH витамина D и биогенных аминов в различные фотопериоды года у населения Архангельска, а также в дальнейшем выявлении типологических вариантов внутрииндивидуальной динамики изучаемых показателей в зависимости от пола и продолжительности светового дня.
ЦЕЛЬ. Оценить сезонную обеспеченность витамином D и биогенными аминами у практически здоровых мужчин трудоспособного возраста, проживающих в городе Архангельске.
МЕТОДЫ. Проведено аналитическое проспективное неконтролируемое исследование с участием 20 клинически здоровых мужчин города Архангельска (64°32′24,4″с.ш.). Образцы крови собраны в каждый сезон (март, июнь, сентябрь, декабрь) в течение года. Определены концентрации витамина D, серотонина, дофамина, адреналина и норадреналина в крови. Статистический анализ эндокринных показателей проводили с помощью рангового критерия Вилкоксона с применением поправки Бонферрони.
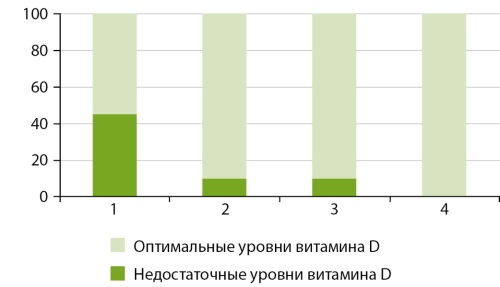
РЕЗУЛЬТАТЫ. Установлена годовая динамика концентрации 25-ОН Витамина D в крови у мужчин с минимальным его уровнем в период увеличения продолжительности светового дня (март) с последующим повышением концентрации витамина летом (июнь) и максимальным уровнем в период снижения продолжительности светового дня (сентябрь). В период увеличения светового дня (март) отмечается наибольший процент лиц (45%) с недостаточной (ниже 30 нг/мл) концентрацией витамина D в крови, при этом в летний и осенний период этот процент снижается до 10, а зимой недостаточные концентрации витамина D не обнаружены.
ЗАКЛЮЧЕНИЕ. Наиболее критическим сезоном по содержанию витамина D в крови у мужчин является период увеличения продолжительности светового дня (март), а более благоприятным — период снижения продолжительности светового дня (сентябрь). Сезонная динамика уровня серотонина ассоциирована с изменениями концентраций витамина D в течение года. Сезонные вариации содержания дофамина, адреналина и норадреналина были сходны между собой и отличались от годовой динамики витамина D в крови.
КЛИНИЧЕСКИЕ СЛУЧАИ
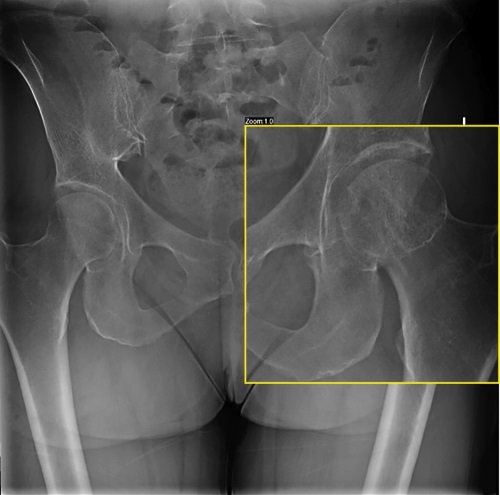
Гипофосфатазия (HPP) — наследственное метаболическое заболевание, характеризующееся низкой активностью ткань-неспецифической щелочной фосфатазы (TNAP) вследствие мутаций в гене ALPL [1]. Низкая активность TNAP приводит к системному накоплению его субстратов, а именно неорганического пирофосфата (PPi), сильного ингибитора минерализации, и пиридоксаль-5’-фосфата (PLP), кофактора нескольких ферментов, которые в значительной степени объясняют скелетно-мышечные и системные особенности заболевания. Гипофосфатазия характеризуется широким спектром проявлений и различной степенью тяжести: от бессимптомного течения у носителей мутации ALPL до внутриутробной смерти при перинатальной форме. В данной статье будет рассмотрена манифестация заболевания во взрослом возрасте, которая требует дифференциальной диагностики с постменопаузальным остеопорозом.
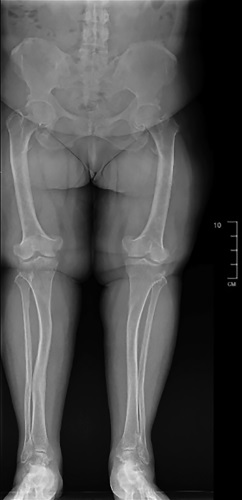
Остеомаляция — это системное заболевание скелета, характеризующееся нарушением минерализации или дефектной минерализацией вновь образованного костного матрикса у взрослых. Наиболее частой причиной является выраженный дефицит витамина D любой этиологии, дефицит кальция и дефицит фосфора (патология почек, мезенхимальные опухоли, секретирующие фактор роста фибробластов 23 (ФРФ23/FGF23), генетические заболевания). Среди генетических заболеваний чаще всего встречается X-сцепленный доминантный гипофосфатемический рахит (ген PHEX, лат. X-linked hypophosphatemic rickets (XLHR), OMIM: 307800), гораздо реже — аутосомно-доминантный гипофосфатемический рахит (ген FGF23, лат. Autosomal dominant hypophosphatemic rickets (ADHR), OMIM: 193100) и аутосомно-рецессивный гипофосфатемический рахит 1 и 2 типа (ген DMP1, ENPP1, FAM20C, лат. Autosomal recessive hypophosphatemic rickets-1 (ARHR1), OMIM: 241520; Autosomal recessive hypophosphatemic rickets-2 (ARHR2), OMIM: 613312). ADHR — крайне редкая наследственная патология, обусловленная мутацией в гене FGF23, для которой характерна манифестация в любом возрасте. В литературе описано около 50 случаев данной патологии. В статье впервые в Российской Федерации представлен клинический случай взрослой пациентки с ADHR. В ходе дифференциальной диагностики такие причины остеомаляции, как выраженный дефицит витамина D, заболевания, связанные с нарушением канальцевой реабсорбции почек, ФРФ23-продуцирующая опухоль, были исключены. Пациентке проведено генетическое исследование, по результатам которого выявлена мутация в гене FGF23, подтвержден диагноз ADHR. Инициирована терапия препаратами фосфора и активными метаболитами витамина D, на фоне которой отмечалось улучшение самочувствия в виде уменьшения болей при ходьбе, увеличения мышечной силы.
Трудности диагностики остеомаляции обусловлены отсутствием рутинного определения уровня фосфора в крови и малой осведомленностью врачей об этом заболевании. В свою очередь генетическое исследование в ряде случаев позволяет подтвердить наследственные формы, что предотвращает неоправданные хирургические вмешательства, обеспечивает своевременное назначение медикаментозной терапии и значимо улучшает качество жизни пациентов.
Гиперкальциемия — это синдром, подтвержденный лабораторно при повышении уровня кальция крови выше 2,55 ммоль/л. Ряд авторов утверждает, что наиболее частыми причинами гиперкальциемии являются злокачественные новообразования, первичный гиперпаратиреоз (ПГТП), интоксикация витамином D, хроническая болезнь почек (ХБП). Одной из наиболее редких причин, которые следует учитывать у пациентов с впервые диагностированной гиперкальциемией, является синдром семейной гипокальциурической гиперкальциемии (СГГ). СГГ является генетическим аутосомно-доминантым заболеванием, характеризующимся сниженным уровнем кальция мочи и повышенным уровнем кальция крови в сочетании с нормальным или превышающим референсные значения уровнем ПТГ. Представленный случай демонстрирует необходимость выполнения дифференциальной диагностики синдрома гиперкальциемии, важность расчета отношения почечного клиренса кальция к клиренсу креатинина, что в настоящее время является самым доступным методом и позволяет избежать необоснованного выполнения паратиреоидэктомии.
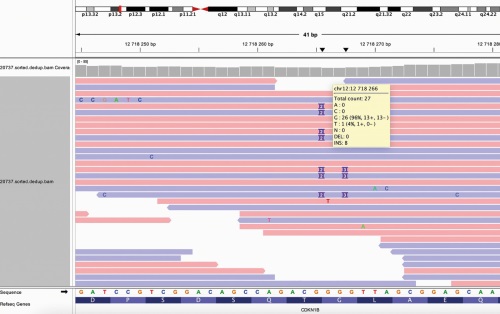
Синдром множественных эндокринных неоплазий 4 типа (МЭН-4) — редкое аутосомно-доминантное заболевание, возникающее в результате мутации в гене CDKN1B, кодирующем регулятор клеточного цикла p27. В связи с редкостью патологии к настоящему времени собрано небольшое количество клинических наблюдений за пациентами с мутациями CDKN1B, при этом наличие генотип-фенотипических корреляций остается дискутабельным и требует дополнительного уточнения. При МЭН-4 в патологический процесс вовлекаются те же органы, что и при МЭН-1, однако возраст манифестации и течение заболевания могут отличаться. Мы представляем описание клинического случая пациентки с МЭН-4 с ранее не описанной в литературе мутацией в гене CDKN1B со сдвигом рамки считывания, которая привела к развитию первичного гиперпаратиреоза (ПГПТ) с множественным поражением околощитовидных желез (ОЩЖ) и пролактин-секретирующей микроаденомы гипофиза. Первым компонентом заболевания, диагностированным в возрасте 37 лет, была пролактинома. Позднее была выявлена висцеральная форма ПГПТ с изолированным поражением почек. Пациентке проводилось специфическое лечение указанных патологий с достижением удовлетворительных результатов. При комплексном обследовании значимого поражения других органов выявлено не было. Ограниченное число наблюдений за пациентами с мутациями в гене CDKN1B в настоящее время не позволяет определить закономерности течения этого заболевания, в связи с чем подробное описание клинической картины при новой выявленной мутации вносит значимый вклад в изучение данной патологии.
Контент доступен под лицензией Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2311-0716 (Online)
































