



CLINICAL GUIDELINES
BACKGROUND: The Arctic zone territories of the Russian Federation have the risk of developing 25-OH vitamin D deficiency, which is aggravated by low levels of insolation and nutrition low in vitamins and minerals. Along with low levels of insolation and reduced vitamin D concentrations during periods of minimal daylight hours, manifestations of seasonal affective disorder may occur, which is characterized by circadian dysregulation and monoaminergic functions of neurotransmitters (serotonin, dopamine, noradrenaline). The scientific challenge lies in estimating 25-OH vitamin D and biogenic amines sufficiency and detecting associative changes in 25-OH vitamin D and biogenic amines levels during various photoperiods of the year in the population of Arkhangelsk, as well as in further identification of typological variations in the intra-individual dynamics of the studied indicators depending on gender and daylight hours.
AIM. The purpose is to assess the seasonal sufficiency of vitamin D and biogenic amines for apparently healthy men of working age living in the city of Arkhangelsk.
METHODS. An analytical prospective uncontrolled study was conducted with the participation of 20 clinically healthy men from the city of Arkhangelsk (64°32’24.4’’N). Blood samples were collected every season (March, June, September, December) during the year with subsequent determination of the concentrations of vitamin D, serotonin, dopamine, adrenaline and noradrenaline in the blood. Statistical analysis of endocrine parameters was performed using the Wilcoxon rank test with the Bonferroni correction.
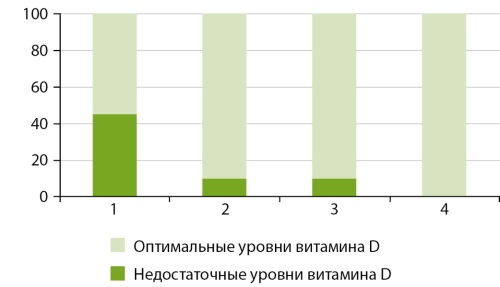
RESULT: According to the revealed annual dynamics, the concentration of 25-OH vitamin D in the blood of men who live in the city of Arkhangelsk reached a minimum level during an increase in daylight hours (March), followed by an increase in vitamin concentration in summer (June) and a maximum concentration level during a decrease in daylight hours (September). The largest percentage of people (45%) with insufficient (below 30 ng /ml) vitamin D concentration in the blood is noted during the period of increasing daylight (March), while in summer and autumn this percentage decreases to 10, and in winter no insufficient concentrations of vitamin D are detected.
CONCLUSION: In terms of vitamin D concentration in the blood of men, the period of increase in daylight hours (March) is the most critical season, whereas the period of decrease in daylight hours (September) is a more favourable one. Seasonal dynamics of serotonin concentrations were associated with changes in vitamin D levels during the year. Seasonal variations of dopamine, adrenaline and noradrenaline concentrations were similar to each other and different from the annual dynamics of vitamin D concentration in the blood.
CASE REPORTS
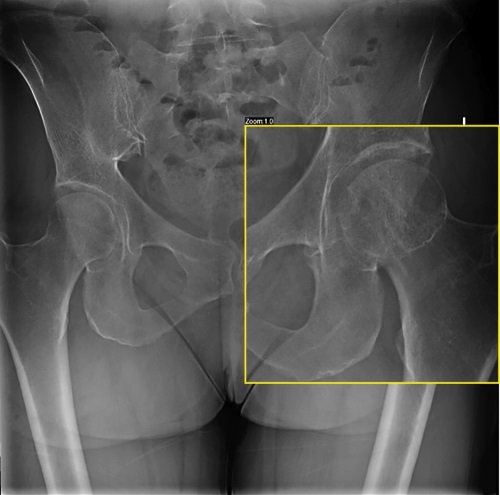
Hypophosphatasia (HPP) is an inherited metabolic disease characterized by low activity of tissue non-specific alkaline phosphatase (TNAP) due to mutations in the ALPL gene [1]. The low activity of TNAP leads to the systematic accumulation of its substrates, namely inorganic pyrophosphate (PPi), a strong inhibitor of mineralization, and pyridoxal-5’-phosphate (PLP), a cofactor of several enzymes, which largely explain the musculoskeletal and systemic features of the disease. Hypophosphatasia is characterized by a wide range of manifestations and varying degrees of severity: from asymptomatic course in carriers of the ALPL mutation to intrauterine death in perinatal form. This article will discuss the manifestation of the disease in adulthood, which requires differential diagnosis with postmenopausal osteoporosis.
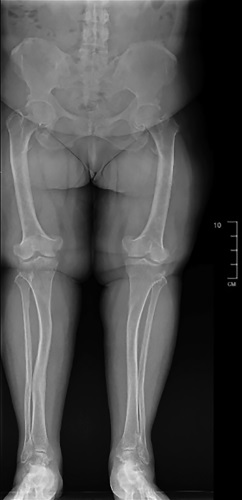
Osteomalacia is a systemic disease of the skeleton, accompanied by the formation of an unmineralized or poorly mineralized osteoid instead of full-fledged bone tissue. The most common cause is severe vitamin D and calcium deficiency, phosphorus deficiency (kidney pathology, mesenchymal tumors secreting an excess of FGF23, genetic diseases). Among inherited pathologies, X-linked dominant hypophosphatemic rickets (XLHR, gene PHEX, OMIM: 307800) is the most frequent form, while autosomal dominant hypophosphatemic rickets (ADHR, gene FGF23, OMIM: 193100) and autosomal recessive hypophosphatemic rickets 1,2 (ARHR1-2, genes DMP1, ENPP1, FAM20C, OMIM: 241520, OMIM: 613312) are much less common. ADHR is an extremely rare form of genetic rickets caused by mutations in the FGF23 gene. It can manifest at any age. About 50 cases of this disease have been reported in the literature. This article presents the first clinical case of ADHR in an adult in the Russian Federation. Severe vitamin D deficiency, renal tubular disorders and tumor-induced osteomalacia were excluded in differential diagnosis. The patient underwent a genetic test, which revealed a mutation in the FGF23 gene and confirmed the diagnosis of ADHR. Therapy with an active vitamin D analog and phosphate supplement was initiated, after which the patient noticed decreased pain when walking and increased muscle strength.
Difficulties in diagnosing osteomalacia are due to the lack of routine determination of serum phosphate and low awareness of doctors about this disease. In some cases, genetic tests make it possible to confirm hereditary forms, which prevents unnecessary surgical treatment, ensures timely prescription of therapy and significantly improves the quality of patients’ lives.
Hypercalcemia is a laboratory-confirmed syndrome with an increase in blood calcium levels above 2.55 mmol/l. A number of authors claim that the most common causes of hypercalcemia are malignant neoplasms, primary hyperparathyroidism (PHPT), vitamin D intoxication, and chronic kidney disease. One of the rarest causes to consider in patients with newly diagnosed hypercalcemia is familial hypocalciuric hypercalcemia syndrome (FHH). FHH is a genetic autosomal dominant disorder characterized by decreased urinary calcium levels and increased blood calcium levels in combination with normal or above-reference PTH levels. The presented case demonstrates the need to perform a differential diagnosis of hypercalcemia syndrome, the importance of calculating the ratio of renal calcium clearance to creatinine clearance, which is currently the most accessible method and allows one to avoid unjustified parathyroidectomy.
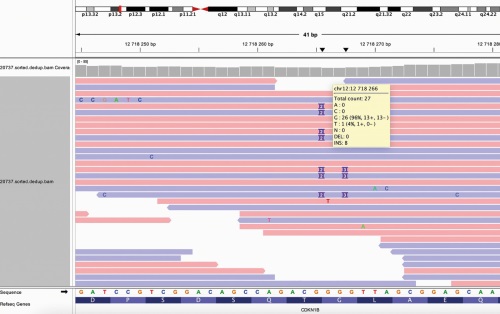
Multiple endocrine neoplasia syndrome type 4 (MEN-4) is a rare autosomal dominant disease caused by mutation in the CDKN1B gene encoding the cell cycle regulator p27. Currently, a small number of clinical cases of patients with this pathology are known. Patterns of genotype-phenotype correlations in patients with CDKN1B mutations remains controversial and requires additional clarification. MEN-4 affects the same organs as MEN-1, however, the age of manifestation and the course of the disease may differ. We present the clinical case of a patient with MEN-4 with a new frame shift mutation in the CDKN1B gene caused PHPT with multiple parathyroid gland pathology and prolactin-secreting pituitary microadenoma. The first component of the disease diagnosed at the age of 37 was prolactinoma. Later, a visceral form of PHPT with isolated kidney complication was revealed. The patient has got the necessary treatment of the identified pathologies with satisfactory results. During a comprehensive examination, the involvement of other endocrine organs was not revealed. The limited number of case reports of patients with mutations in the CDKN1B gene currently does not allow us to determine the patterns of this syndrome’s course, and therefore a detailed description of the disease’s clinical presentation in a patient with a newly identified mutation makes a significant contribution to the study of this pathology.
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2311-0716 (Online)
































