



Original study
BACKGROUND: Osteogenesis imperfecta (Q78.0) and Ehlers-Danlo syndrome (Q79.6) are rare inherited (orphan) connective tissue diseases with variable clinical manifestations and a wide range of molecular defects. Data on the prevalence of these diseases are fragmentary due to the heterogeneous clinical picture and low availability of molecular diagnostics both in the world and in Russia. However, understanding the frequency of these diseases may help to optimize diagnosis and improve the quality of medical care for this category of patients.
AIM: To analyze the prevalence of osteogenesis imperfecta and Ehlers-Danlo syndrome in the Republic of Bashkortostan according to medical records.
MATERIALS AND METHODS: Data on the population of the republic were obtained from the official website of the Federal State Statistics Service for the Republic of Bashkortostan. Information on the incidence of diseases was taken from the reports of the Medical Information and Analytical Center of the Republic of Bashkortostan, analysis of the card file of the Republican Medical and Genetic Center (Ufa) and analysis of the Republican Medical Information and Analytical System «ProMed». Molecular genetic studies were performed by massively parallel sequencing.
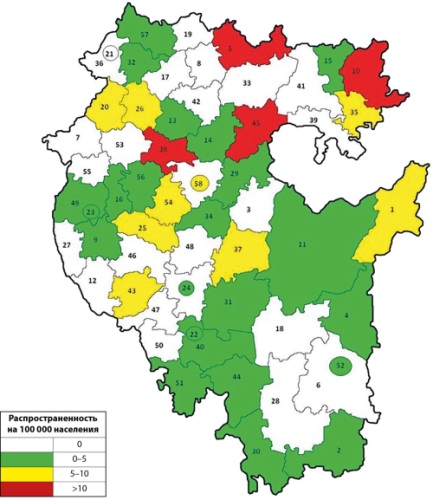
RESULTS: A total of 199 patients with osteogenesis imperfecta have been identified in the Republic of Bashkortostan, 69 (34.6%) of them are under active observation. Molecular genetic study was performed for 64 patients (32.1%), and 16 pathogenic changes in the COL1A1 gene, 11 pathogenic mutations in the COL1A2 gene, one mutation each in the P3H1 and IFITM5 genes were detected. 112 males and 87 females were identified. The share of patients under 18 years of age in the whole republic amounted to 36.1%. In Ufa 80 patients were registered, the percentage of patients under active surveillance amounted to 42.5%. In general, 133 patients with Ehlers-Danlo syndrome were registered in the Republic of Bashkortostan, of which 30 patients (22.5%) were under active surveillance. Molecular genetic study was carried out in 43 patients (32.3%), and 5 changes of nucleotide sequence in the COL5A1 gene and 3 in the COL5A2 gene were revealed. There were 71 men and 62 women. As in the case of LE, patients older than 18 years prevail — 89 patients (66.9%), 18 patients (28.6%) were under active surveillance. The overall prevalence of osteogenesis imperfecta in the Republic of Bashkortostan amounted to 5.031 cases per 100,000 population, Ehlers-Danlo syndrome — 3.362 cases per 100,000 population. Patients with osteogenesis imperfecta are more often on active follow-up compared to Ehlers-Danlo syndrome (48.6% and 22.5%, respectively), which is explained by a longer period of work with this group of patients.
CONCLUSION: The prevalence of osteogenesis imperfecta and Ehlers-Danlo syndrome in the Republic of Bashkortostan according to medical records was determined. The total prevalence of osteogenesis imperfecta in the Republic of Bashkortostan amounted to 5.031 cases per 100 thousand population, Ehlers-Danlo syndrome - 3.362 cases per 100 thousand population.
BACKGROUND: Systemic lupus erythematosus (SLE) is a chronic disease of unknown etiology characterized by systemic immuno-inflammatory damage to vital organs. A serious complication of SLE is aseptic bone necrosis, which is characterized by severe pain syndrome, impaired joint function, decreased quality of life and disability. Despite the fact that hip replacement is a technically proven method of surgical treatment, patients with SLE are still at increased risk of complications at all stages of the perioperative period.
AIM: the aim of the work is to describe the effect of SLE on the development of intraoperative complications during hip replacement.
MATERIALS AND METHODS: The study included 133 patients diagnosed with SLE who underwent total hip replacement between 1998 and 2021. All surgical interventions were performed in the traumatology and orthopaedic department. The group of patients with SLE was dominated by women, the ratio of women to men was 7.3:1, respectively. The average age of patients at the time of surgery was 36.6±12.6 years.
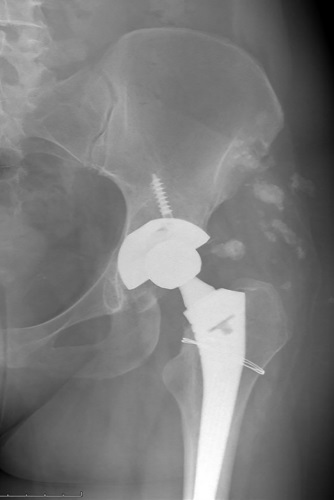
RESULTS: Hip replacement in all patients (n=133) was performed as standard in the patient’s position on the healthy side from the anterolateral Harding access. In 75 cases (56.3%), additional fixation of the acetabulum component with screws was performed. In 10 cases, a cerclage was applied to the proximal femur due to a fracture that occurred during the development of the bone marrow canal. To clarify the factors influencing the development of an intraoperative fracture, patients with SLE were divided into 2 groups: the first group (n=10) — with an intraoperative fracture, the second (n=123) — without a fracture. The comparative characteristics of the first and second groups are carried out. Another complication at the stage of surgical treatment is an increased volume of blood loss, which required transfusion of erythrocyte suspension. There were 54 such patients (40.6%). In relation to patients with increased blood loss, we compared the clinical and laboratory characteristics of patients immediately before surgical treatment, with and without increased blood loss.
CONCLUSION: In the study, we analyzed the resulting intraoperative complications. We have not received confirmation of the effect of daily and cumulative doses of HA, as well as the degree of activity and course of SLE on the risk of intraoperative fracture. The probability of fracture was significantly higher in patients with initially reduced bone mineral density. With regard to the increased volume of blood loss in patients with SLE, a significant association was found with baseline anemia (reduced hemoglobin and hematocrit levels) in the preoperative period (p<0.001).
BACKGROUND: Diabetic peripheral polyneuropathy (DPPN) is a common and early chronic complication of diabetes mellitus (DM). This complication can be associated with bone tissue damage and low-trauma fractures, particularly in type 1 DM. Therefore, evaluating the relationship between DPPN and bone metabolism parameters is crucial for clinical practice.
AIM: To assess bone remodeling parameters in patients with type 1 DM with varying degrees of DPPN.
MATERIALS AND METHODS: The study included 90 patients with type 1 DM and DPPN aged 18 to 55 years on stable insulin therapy for the past 3 months. Assessment was conducted for levels of HbA1c, 25(OH)D, total calcium (Ca), phosphorus (P), parathyroid hormone (PTH), osteocalcin (OC), C-terminal telopeptide of type I collagen (CTX), fibroblast growth factor 23 (FGF-23) in serum. The degree of neurological deficit was determined using the Neuropathy Disability Score (NDS), Neuropathy Symptoms Score (NSS), and Michigan Neuropathy Screening Instrument (MNSI). Dual-energy X-ray absorptiometry (DXA) was performed to determine bone mineral density (BMD) and trabecular bone score (TBS).
RESULTS: Regardless of the degree of DPPN severity, patients with type 1 DM were comparable in HbA1c and 25(OH)D levels. A direct correlation was found between the severity of neurological disorders and BMI (r=0.214; p=0.044) and duration of DM (r=0.246, p=0.019). Vitamin D insufficiency and deficiency were diagnosed in 79% of examined patients with type 1 DM, but the level of 25(OH)D in serum did not depend on the degree of DPPN severity, HbA1c level, duration of DM, and daily insulin dose (p>0.05). A positive correlation was established between PTH level and severity of neurological symptoms, and a negative one with HbA1c level. Patients with type 1 DM and varying degrees of DPPN did not differ in BMD and TBS. A Z-score of less than -2.0 SD was only recorded in six patients, and partially degraded microarchitecture of bone tissue was observed in 8 patients.
CONCLUSION: Therefore, BMD, TBS, and bone remodeling markers in patients with type 1 DM and DPPN were independent of the degree of neurological deficit, degree of compensation, and duration of DM. Vitamin D deficiency is common in most patients with type 1 DM, but the 25(OH)D level is not influenced by HbA1c level, duration of the disease, and daily insulin dose.
Review
Hereditary skeletal dysplasias (HSD) are primary growth disorders; occur with a frequency of 1: 5000 newborns; characterized by wide phenotypic heterogeneity. Diagnosis of НSD is based on clinical symptoms (dyspropotion of body segments), radiographic characteristics of bone mineralization, maturation and morphology, and molecular genetic studies. Treatment of this group of patients must be multidisciplinary, since it affects not only skeleton. The role of the pediatric endocrinologist is to assess the growth potential of these patients and suggest the most optimal treatment. The review provides data on various types of spondyloepi(meta)physeal dysplasias (SE(M)D), a large heterogeneous group of HSD associated with short stature. Clinical manifestations of SEMD include short stature, disorders in development of spondyles, epiphyses and metaphyses of tubular bones, advanced bone age, etc. It is necessary to differentiate the most common causes of short stature from genetic skeletal diseases, which are characterized by wide phenotypic heterogeneity and require careful examination. In this review, we analyse the literature data on spondyloepiphyseal dysplasia, as one of the causes of idiopathic short stature in children.
Case report
Neurofibromatosis type 1 (NF-1) is an autosomal dominant disorder caused by mutations in NF1 gene encoding neurofibromin protein. Mutations in this gene result in the absence of suppression of the Ras-MAPK pathway, which leads to increased cell proliferation and tumor development. In addition to the classic manifestations of neurofibromatosis (skin manifestations, tumors of the peripheral and cranial nerves, and scoliosis), patients may develop tumors of the endocrine glands: pheochromocytomas/paragangliomas and neuroendocrine tumors. Cases of NF-1 combined with acromegaly, adrenal tumors (including adrenocortical cancer), and primary hyperparathyroidism (PHPT) have been described. In addition, NF-1 is notable for the development of hypophosphatemic osteomalacia in rare cases.
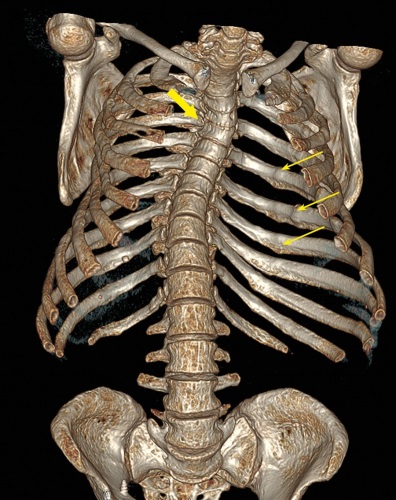
The article presents a description of a clinical case of a combination of PHPT in a patient with NF-1, hypophosphatemia that persisted after successful surgical treatment of PHPT, and severe skeletal involvement with multiple vertebral and non-vertebral fractures, which was assessed as a combination of severe osteoporosis and hypophosphatemic osteomalacia. During treatment with denosumab, teriparatide and alfacalcidol, increase of bone mineral density and normalization of phosphate levels were achieved. Nevertheless, the absence of consolidation of the femoral shaft fracture persists. Few cases of a combination of NF-1 simultaneously with PHPT and hypophosphatemic osteomalacia are described in the literature.
This work is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License (CC BY-NC-ND 4.0).
ISSN 2311-0716 (Online)
































